一种菰cDNA文库的构建方法与流程
本发明涉及分子生物学领域,具体地说,涉及一种菰cDNA文库的构建方法。
背景技术:
随着分子生物学及生物信息学技术的迅速发展,挖掘、克隆新基因、研究新基因的功能已成为功能基因组研究中的一项重要工作。构建cDNA文库是功能基因组学研究的基本手段之一,其在研究某类特定细胞中基因组的表达状态及表达基因的功能鉴定具有特殊的优势,在个体发育、细胞分化、细胞周期调控、细胞衰老和凋亡等生命现象的研究中具有更为广泛的应用价值。然而,以常规方法构建的cDNA文库面临的最大问题是高通量分析过程中的重复拷贝比例高,其主要原因在于高等真核生物的细胞中基因表达水平的差异较大。高拷贝基因序列的大量存在给基因的筛选和鉴定带来了不必要的浪费。自1990年以来出现了各种均一化cDNA文库的构建方法,试图在文库测序前降低高丰度转录本的克隆,增加中、低丰度转录本出现的频率,以有利于低丰度表达基因的发现。
CN102797044A公开了一种高效快速的均一化全长cDNA文库构建方法,该方法利用特定的5-OH寡核苷酸封闭非全长mRNA,在用烟草酸焦磷酸酶除去mRNA5′端的帽子结构,使mRNA5′端帽子结构处的磷酸基团暴露出来,用高效的RNA连接系统在mRNA的5′端连上一段优化的寡聚核糖核酸作为引发第二链cDNA合成的引物结合位点,最后经反转录,二链合成、分级纯化后,将双链DNA克隆于载体中。该方法具有简洁高效的全长mRNA筛选功能,实现对真核生物全长mRNA的捕获,mRNA5′末端含有生物素标记,可用于全长mRNA分离,所用寡核苷酸引物含有优化的修饰方法及核苷酸序列,可实现真核生物mRNA全场捕获、反转录以及DNA的合成。然而,该方法由于采用了RNaseⅠ切割单链RNA,从而影响全长cDNA筛选的严谨性及cDNA文库的全长比例;同时,磁珠的非特异性吸附性也将影响全长比例。
CN101597630公开了一种双链特异性DNA酶介导的cDNA均一化消减杂交方法,依次包括第一链cDNA的合成、合成双链cDNA、转录RNA、逆转录、均一化和消减杂交、双链特异性DNA酶消化、PCR扩增、克隆差异表达基因及鉴定等步骤。与其它方法相比本发明的有益效果主要体现在:(a)对共同表达基因的高消减效率,对差异表达基因的高效富集,同时表达量很低的差异表达基因可以被有效富集;(b)能够得到全长cDNA序列。然而,该方法增加了消减杂交的步骤,实验方法繁琐、复杂。
罗洪林等为构建大片吸虫成虫cDNA表达文库,用Trizol试剂提取大片吸虫成虫总RNA,经反转录合成cDNA第一链,应用LD-PCR扩增方法,合成双链cDNA。用SfiⅠ内切酶修饰此双链cDNA,使形成两端分别带有SfiⅠA和SfiⅠB的黏性末端。经CHROMA SPIN-400柱纯化,收集400bp以上的双链cDNA片段,将其连接于带有SfiⅠA和SfiⅠB末端的λTriplEx2噬菌体载体,经体外包装后,以XL1-Blue为受体菌构建cDNA表达文库。经测定,库容量为1.08×106PFU/mL,重组率为96.6%。扩增后的文库滴度为2.41×109PFU/mL,插入片段平均大小约为1 000bp。这些结果表明已成功构建大片吸虫成虫cDNA表达文库,适合进一步筛选大片吸虫新基因(应用LD-PCR法构建大片吸虫成虫cDNA表达文库,《畜牧兽医学报》2005年11期)。然而,该方法存在文库非均一化的问题,不利于低丰度表达基因的发现。
菰(Zizania latifolia(Griseb.)Stapf)是国家二级保护植物品种,也是水稻的近缘属,具有生物量大、抗病虫、耐寒等许多优良性状,近年来被众多研究学者作为水稻育种优良基因资源库的重要来源。然而,针对菰的分子生物学研究仍然处于起步阶段,菰的遗传信息获得还很不完整,对菰基因信息的有限所知严重阻碍了其作为水稻育种优良基因资源库的研究与应用。因此开发一种快速、简单、高效的菰cDNA文库的构建方法,解决常规操作步骤复杂,RNA容易降解的问题,克服基因转录水平上的巨大差异,提高发现随机序列和稀有基因的几率,对于开展菰功能基因的利用研究十分必要。
技术实现要素:
为了解决现有技术中存在的问题,本发明的目的是提供一种菰cDNA文库的构建方法。
为了实现本发明目的,本发明的技术方案如下:
一种菰cDNA文库的构建方法,其中,为了提高cDNA反转录效率,本发明通过SMART法,利用反转录酶PowerScript RT的末端转移酶活性来实现cDNA反转录。
进一步地,DSN酶均一化效率的高低是构建均一化cDNA文库的关键,本发明通过对杂交后的cDNA利用不同浓度梯度的DSN进行消化,富集低丰度表达基因,显著提高了DSN酶均一化效率。
具体而言,本发明所述的菰cDNA文库的构建方法包括如下步骤:
S1、菰总RNA获得;
S2、cDNA第一链合成;
S3、LD PCR;
S4、蛋白酶K消化;
S5、cDNA文库均一化:
S51、杂交;
S52、DSN消化;
S6、DSN消化后两次PCR放大;
S7、扩增产物纯化;
S8、Sfi I消化;
S9、酶切cDNA纯化。
进一步地,所述构建方法还包括如下步骤:
S10、cDNA与PUC19载体连接;
S11、质粒的转化;
S12、克隆鉴定。
进一步地,本发明针对菰的特殊性优化了反应体系、反应条件及所用引物序列。
其中,S2具体包括如下步骤:
a.在PCR管中加入以下试剂:
b.72℃温浴2min;
c.冰浴2min,离心后加入下列试剂:
d.42℃温浴1h。
其中,CDS III的序列为:5'-AAGCAGTGGTATCAACGCAGAGTGGCCGAGGCGGCCd(T)20VN*-3';N=A,C,G or T;V=A,G or C。
其中,S52具体包括如下步骤:
a.设置不同浓度的DSN酶:
①将DSN storage buffer和DSN酶等体积混匀,标记为1/2DSN;
②将3倍体积的DSN storage buffer和1倍体积的DSN酶混匀,标记为1/4DSN;
③DSN storage buffer,标记为control;
④DSN酶,标记为DSN;
b.68℃预热DSN master buffer;
c.依次加入下列试剂后,混匀;
d.将4个PCR管68℃反应25min;
e.在各管中加入2×DSN stop buffer,混匀;
f.室温放置5min;
g.-20℃保存。
其中,S6具体包括如下步骤:
S61、第一次放大
a.分别向S52得到的四个消化后产物中加入以下试剂,混匀;
其中,Primer M1为5'-AAGCAGTGGTATCAACGCAGAGT-3’;
b.PCR:95℃1min;95℃15sec、66℃20sec、72℃3min,11cycles;
c.从各管中分别取PCR产物进行电泳检测;
d.根据电泳结果,选择条带大小为1000-2000bp、亮度一致、分散均匀的产物为模板进行第二次放大。
其中,S6具体包括如下步骤:
S62、第二次放大
a.在两个PCR管中依次加入以下试剂,混匀;
其中,Primer M2为5'-AAGCAGTGGTATCAACGCAG-3’;
b.PCR:
c.取PCR产物进行电泳检测。
作为优选,构建过程中所需要的叶片为新鲜、幼嫩的组织,从而保证获得数量足够的高质量的起始RNA。
本发明的有益效果在于:
本发明提供了一种菰cDNA文库的构建方法,以菰的幼嫩叶片作为试验材料,由RNA提取,cDNA第一链合成,LD PCR,蛋白酶K消化,cDNA文库均一化,两次PCR放大,扩增产物纯化,Sfi I消化,酶切cDNA纯化,cDNA与PUC19载体连接,质粒的转化,以及克隆鉴定等12个步骤完成;该方法构建文库便捷,文库容量大,重组率高,完整性好,均一化后高丰度基因明显降低,为功能基因的验证奠定了基础。
本发明构建的菰cDNA文库库容量大,达到了3.6×106pfu·mL-1,插入片段平均长度为1000bp,重组率为97.9%,均一化全长cDNA文库能有效富集低丰度表达基因,降低冗余率。该文库适用于目的基因的筛选,以及后续的基因、蛋白互作分析。
附图说明
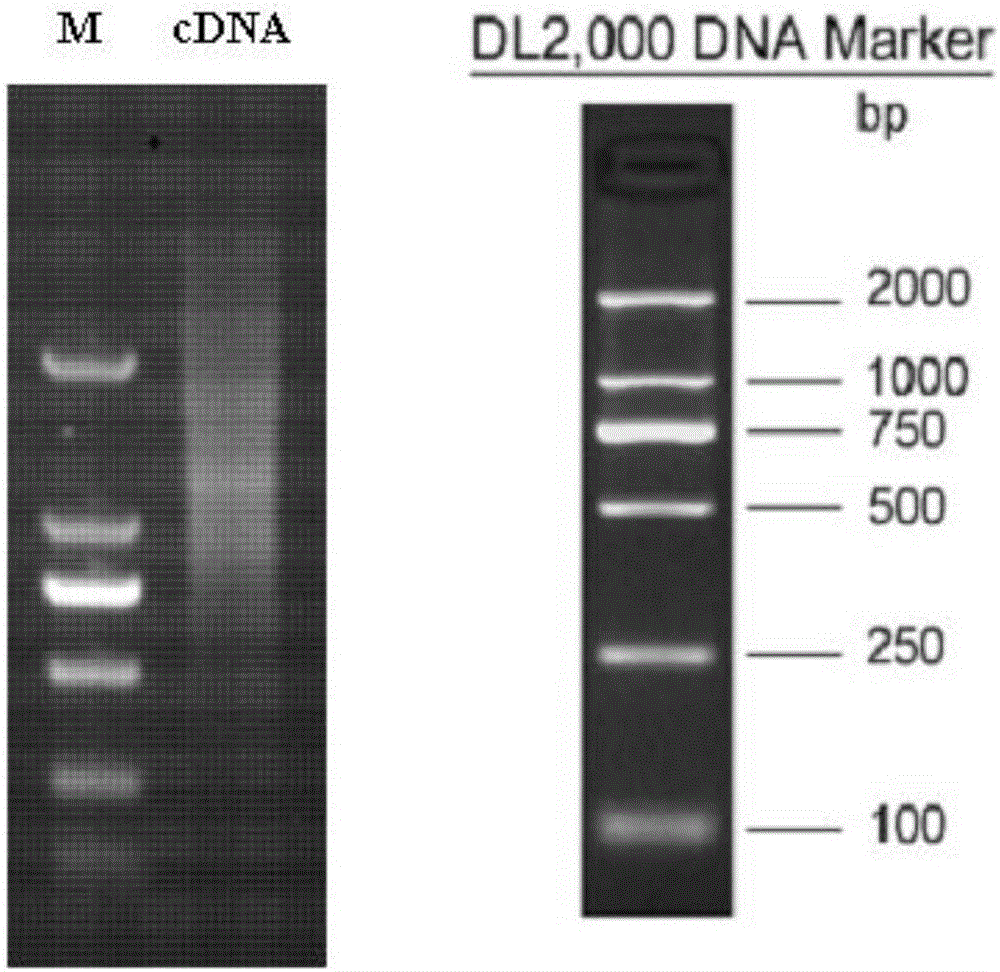
图1为双链cDNA合成后的电泳检测图,其中,M为Marker(TAKARA公司的DL2000)。
图2为均一化后一次放大的电泳检测图,其中,M为Marker(TAKARA公司的DL2000)。
图3为均一化后二次放大的电泳检测图,其中,M为Marker(TAKARA公司的DL2000)。
图4为验证均一化效率的电泳检测图,其中,M代表Marker(TAKARA公司的DL2000),N代表均一化模板,UN代表未均一化模板,C代表cycle数目。
图5为菌落的生长情况。
图6为插入片段鉴定的电泳检测图,其中,M为Marker(TAKARA公司的DL2000),从左至右依次为挑选的第1-24个克隆的PCR产物。
具体实施方式
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1
本发明的菰均一化cDNA文库的构建方法主要包括以下步骤:总RNA的提取,利用SMART方法合成cDNA双链,cDNA变性后重新杂交,经双链特异性核酸酶降解双链cDNA后完成cDNA的均一化过程,均一化后的单链cDNA成分经PCR扩增并插入载体等。
具体步骤如下:
(1)菰总RNA获得:
a.取1g菰的幼嫩叶片,在预冷的研钵中用液氮研成粉末,然后转入15mL RNase free离心管中;
b.加入10mL的Trizol试剂,振荡混匀,室温静置10min充分裂解细胞;
c.加入2mL的氯仿,剧烈震荡15sec,室温静置5min;
d.12,000×g,4℃,离心15min;
e.取上清至新15mL RNase free离心管中,加入等体积异丙醇,颠倒混匀,室温静置5min;
f.12,000×g,4℃,离心10min;
g.弃上清,加入5mL的75%乙醇(RNase free)洗涤沉淀,12,000×g高速冷冻离心5min;
h.重复上述步骤一次;
i.弃上清,超净工作台中空气干燥RNA沉淀,然后溶解于500μLDEPC水中。
(2)cDNA第一链合成
a.在0.2mL PCR管中加入以下试剂:
CDS III/3'PCR Primer:5'-AAGCAGTGGTATCAACGCAGAGTGGCCGAGGCGGCCd(T)20VN*-3';N*=A,C,G or T;V=A,G or C。
b.72℃温浴2min;
c.冰浴2min,离心加入下列试剂:
d.42℃温浴1h。
(3)LD PCR
a.在0.2mL反应管中加入下列试剂:
b.轻拍混匀各组分,稍稍离心后放到已预热(95℃)的PCR仪。按下面程序开始PCR:
图1所示为扩增后cDNA的1%琼脂糖凝胶电泳图,可见200bp-2000bp之间亮度不同的弥散条带,M为分子量标准。
(4)蛋白酶K消化
a.每50μL扩增的双链cDNA中加入2μL蛋白酶K(20μg/μL);混匀并短暂离心;
b.45℃温浴20min,短暂离心;
c.加入等体积酚:氯仿:异戊醇剧烈震荡1-2min;
d.14,000rpm离心5min;
e.收集上层液体到另一干净的离心管中,弃去中间层和下层溶液;
f.加入等体积的氯仿:异戊醇剧烈震荡1-2min;
g.14,000rpm离心5min;
h.收集上层液体到另一干净的离心管中,弃去中间层和下层溶液;
i.加入1/10体积的3M乙酸钠,2.5倍体积95%的室温乙醇,室温条件下立即14,000rpm离心20min;
g.弃上清,用100μL 80%乙醇洗沉淀物;空气干燥沉淀物约10min;
k.加25μL的去离子水溶解沉淀;
l.取5μL样品电泳检测。
(5)cDNA文库均一化
①杂交
a.将下列试剂于0.2mL PCR管中混匀;
b.将上述溶液平均分成4份,置于0.2mL PCR管中;
c.将PCR管在PCR仪上98℃放置2min;
d.然后将PCR管迅速转移到准备好的68℃PCR仪中5h。
②DSN消化
a.在杂交完成前准备好以下浓度的DSN酶:
①将1μL DSN storage buffer和1μL DSN酶混匀,标记为1/2DSN;
②将3μL DSN storage buffer和1μL DSN酶混匀,标记为1/4DSN;
③直接取1μL DSN storage buffer,标记为control;
b.68℃预热DSN master buffer;
c.依次加入下列试剂后,混匀
d.将4个PCR管68℃反应25min;
e.在各管中加入10μL 2×DSN stop buffer,混匀;
f.室温放置5min;
g.-20℃保存。
(6)DSN消化后两次PCR放大
①第一次放大
a.在四个PCR管中依次加入以下试剂,混匀,分别标记为DSNP1,1/2DSNP1,1/4DSNP1,controlP1
Primer M1:5'-AAGCAGTGGTATCAACGCAGAGT-3’;
b.轻拍混匀各组分,按下面程序开始PCR:
c.从各管中分别取5μLPCR产物进行电泳检测;
d.根据电泳结果,选择合适的产物(如1/4DSN)为模板进行第二次放大。
②第二次放大
a.在两个PCR管中依次加入以下试剂,混匀,分别标记为1/4DSNP2,controlP2
Primer M2:5'-AAGCAGTGGTATCAACGCAG-3’;
b.弹指混匀各组分,短暂离心后放到已预热(95℃)的PCR仪,按下面程序开始PCR:
c.从各管中分别取5μL PCR产物进行电泳检测。
从图2可以看出,高拷贝基因1倍酶、1/2酶处理过度,1/4酶处理合适,因此用1/4酶处理进行二次PCR。由图3可以看出,通过二次PCR使一次PCR更集中。
(7)扩增产物纯化
a.加入等体积酚:氯仿:异戊醇剧烈震荡1-2min;
b.14,000rpm离心5min;
c.收集上层液体到另一干净的离心管中,弃去中间层和下层溶液;
d.加入等体积的氯仿:异戊醇剧烈震荡1-2min;
e.14,000rpm离心5min;
f.收集上层液体到另一干净的离心管中,弃去中间层和下层溶液;
g.加入1/10体积的3M乙酸钠,2.5倍体积95%乙醇,室温条件下立即14,000rpm离心20min;
h.倒掉上清,用100μL 80%乙醇洗沉淀物;
i.空气干燥沉淀物约10min;
j.加84μL的去离子水溶解沉淀;
k.取5μL样品电泳检测。
经过DSN消化后的两次PCR放大,对均一化效率进行验证。以1/2DSN和control为模板,用18S管家基因进行PCR,产物经电泳后用Tanon GIS软件分析灰度,验证结果如图4所示,结果显示本文库均一化后比非均一化的丰度下降26倍以上。
(8)Sfi I消化
a.在一1.5mL离心管中加入下列试剂:
b.充分混匀,50℃温浴2h;
(9)酶切cDNA纯化
a.加入等体积酚:氯仿:异戊醇剧烈震荡1-2min;
b.14,000rpm离心5min;
c.收集上层液体到另一干净的离心管中,弃去中间层和下层溶液;
d.加入等体积的氯仿:异戊醇剧烈震荡1-2min;
e.14,000rpm离心5min;
f.收集上层液体到另一干净的离心管中,弃去中间层和下层溶液;
g.加入1/10体积的3M乙酸钠,2.5倍体积95%的室温乙醇,室温条件下立即14,000rpm离心20min;
h.倒掉上清,用100μL 80%乙醇洗沉淀物;
i.空气干燥沉淀物约10min;
j.加100μL去离子水溶解沉淀。
(10)cDNA与PUC19载体连接
a.反应管中加入下列试剂:
b.4℃反应过夜。
(11)质粒的转化
a.取200μL感受态宿主菌(DH-5α),放置冰上融化;
b.加入上述1μL连接液,轻轻混匀后置于冰上30min;
c.42℃水浴热休克90sec,迅速放入冰上2min;
d.加入300μL SOC培养基(不含抗生素),置于37℃摇床,转速200rpm,复苏45min;
e.菌液涂布于15cm培养皿上(Apr-IPTG/x-gal LB固体培养基),37℃过夜。
由图5可知,菌落总数为2440,其中蓝斑50;重组率为:(克隆总数-蓝斑总数)/克隆总数,重组率为(2440-50)/2440=97.9%。
(12)克隆鉴定:
a、挑24个克隆,37℃,200rpm培养过夜,取1μL菌液作为PCR模板。在PCR管中依次加入以下试剂,混匀:
M13F:5’-GTAAAACGACGGCCAG-3’;
M13R:5’-CAGGAAACAGCTATGAC-3’。
b、轻拍混匀各组分,稍稍离心后放到PCR仪。按下面程序开始PCR:
c、从各管中分别取5μL PCR产物进行电泳检测。
由图6的结果可知,插入片段多大于750bp且分布在0.75-2.0k之间,符合建库标准。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
序列表
<110> 广东菰稻农业有限公司
<120> 一种菰cDNA文库的构建方法
<130> KHP161118838.2
<160> 5
<170> PatentIn version 3.5
<210> 1
<211> 58
<212> DNA
<213> 人工序列
<220>
<221> misc_feature
<222> (57)..(57)
<223> n is a, c, g, or t
<220>
<221> misc_feature
<222> (58)..(58)
<223> v is a, g, or c
<400> 1
aagcagtggt atcaacgcag agtggccgag gcggcctttt tttttttttt ttttttnv 58
<210> 2
<211> 23
<212> DNA
<213> 人工序列
<400> 2
aagcagtggt atcaacgcag agt 23
<210> 3
<211> 20
<212> DNA
<213> 人工序列
<400> 3
aagcagtggt atcaacgcag 20
<210> 4
<211> 16
<212> DNA
<213> 人工序列
<400> 4
gtaaaacgac ggccag 16
<210> 5
<211> 17
<212> DNA
<213> 人工序列
<400> 5
caggaaacag ctatgac 17
- 还没有人留言评论。精彩留言会获得点赞!