雷替曲塞的溶剂化物及其制备方法与流程
本发明属于药物化学领域,具体涉及一种雷替曲塞的溶剂化一水合物及其制备方法。
背景技术:
雷替曲塞,是由英国的royalmarsden医院的癌症研究署与zeneca共同合作发展的药物。它是一种胸腺合成酶抑制剂,属于叶酸的衍生物,用于治疗晚期直肠结肠癌患者。1996年英国首次上市,商品名tomudex,其结构式如下:
现阶段,对于雷替曲塞晶型报道很少,更多的是集中在对于其制备以及精制方法的公开报道。
原研专利us4992550中公开了碱溶酸析的精制方法,最终制备得到了一种粉末状固态物质,为一水合物晶型,并提供了相关熔点数据180-184℃。目前工业上也大多采用这种碱溶酸析的精制方法,但是该制备方法容易在成品中导致氯化钠的残留。
专利cn201210260633.7、cn201110361869.5以及cn201410844732.9中均公开一种将雷替曲塞溶于有机溶剂后再进行洗涤的重结晶方法,其中,在cn201210260633.7中还提供了一种雷替曲塞的新晶型,但是上述精制方法所形成的中间体容易发粘或者形成凝胶,不利于进行下一步过滤工艺。
技术实现要素:
本发明的目的在于提供一种雷替曲塞的溶剂化物,其作为精制反应的中间产物,为制备高纯度的雷替曲塞提供了另一种路径,由该精制方法制备的目标产物,纯度高、在长期稳定性方面得到了显著的提升,提升了雷替曲塞的药用标准,且该精制工艺简单,适合工业化大生产。
本发明提供一种雷替曲塞的溶剂化合物,其特征在于,结构如下所示:
r=c1-c10的烷基,其中优选r为甲基、乙基中的一种;
优选的雷替曲塞甲醇水合物ⅱ,其x射线粉末衍射图在2θ值为7.9±0.2°、10.2±0.2°、12.9±0.2°、15.2±0.2°、17.0±0.2°°、20.4±0.2°、23.2±0.2°、24.00±0.2°、27.6±0.2°处具有特征峰。优选地,2θ值在7.9±0.2°、10.2±0.2°、12.9±0.2°、15.2±0.2°、17.0±0.2°、18.9±0.2°、19.3±0.2°、20.4±0.2°、23.2±0.2°、24.00±0.2°、24.5±0.2°、25.3±0.2°、27.0±0.2°、27.6±0.2°、30.8±0.2°处具有特征峰。更优选地,xrpd图谱见图1。
本发明提供的雷替曲塞甲醇水合物ⅱ其差示扫描量热(dsc)曲线在48.2℃、98.2℃、178.5℃处有吸热峰,其在48.2℃失去溶剂甲醇分子、98.2℃时失去溶剂水分子,178.5℃为溶化峰。dsc图谱详见图2。
本发明经卡尔费休水分仪测得水分含量在3.46%,约含有一分子水。
本发明提供的雷替曲塞甲醇水合物ⅱ其热重(tg)曲线在加热至110℃时,具有9.5%的失重,由热重数据分析得一分子雷替曲塞中含有一个水分子和一个甲醇分子。热重分析图如图3所示。
进一步,本发明还提供雷替曲塞甲醇水合物ⅱ的单晶,证明其晶型ⅱ中一分子雷替曲塞含有一分子水与一分子甲醇,将其单个晶体接受单晶x射线衍射分析,x单晶衍射图谱如图4,其为单斜晶系,轴长
本发明还提供雷替曲塞乙醇水合物ⅲ,其x射线粉末衍射图在2θ值为7.7±0.2°、10.1±0.2°、12.7±0.2°、14.9±0.2°、16.7±0.2°、19.0±0.2°、20.3±0.2°、22.9±0.2°、23.8±0.2°、24.0±0.2°、27.2±0.2°处具有特征峰。优选地,2θ值在7.7±0.2°、8.1±0.2°、10.1±0.2°、10.4±0.2°、10.8±0.2°、12.7±0.2°、13.2±0.2°、14.9±0.2°、16.7±0.2°、17.5±0.2°、19.0±0.2°、19.8±0.2°、20.3±0.2°、22.9±0.2°、23.8±0.2°、24.0±0.2°、27.2±0.2°、30.6±0.2°、32.5±0.2°处具有特征峰。更优选地,xrpd图谱见图5。
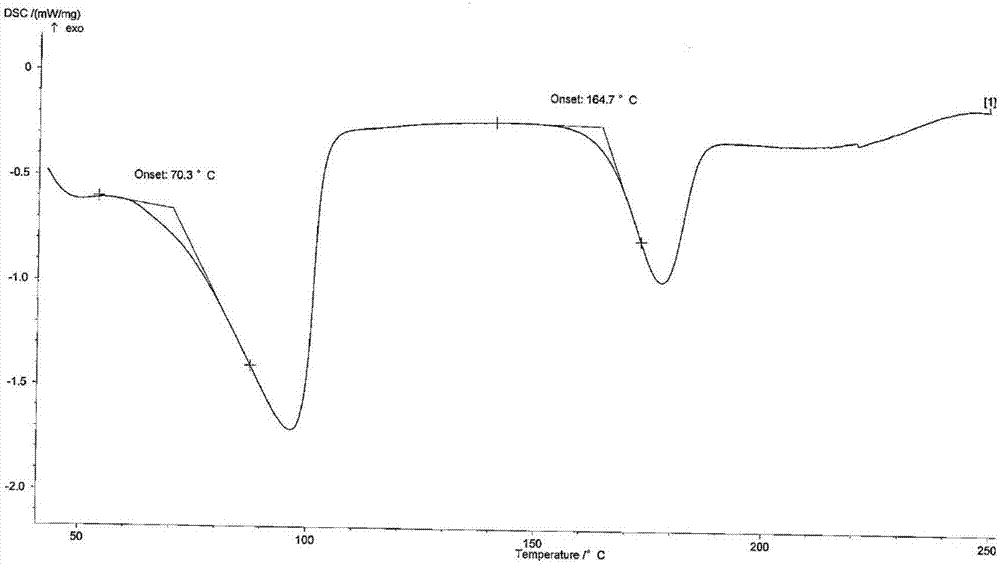
本发明提供雷替曲塞乙醇水合物ⅲ其差示扫描量热法(dsc)曲线在50.0℃、98.2℃、173.5℃处有吸热峰,在50.0℃失去溶剂乙醇分子、98.2℃时失去溶剂水分子,173.5℃为溶化峰。dsc图谱见图6。
本发明经卡尔费休水分仪测得水分含量在3.50%,约含有一分子水。
本发明提供的雷替曲塞乙醇水合物ⅲ其热重(tg)曲线在加热至140℃时,具有12.33%的失重,由热重数据分析得一分子雷替曲塞中含有一个水分子和一个乙醇分子。热重分析图如图7所示。
另一方面,本发明还提供了雷替曲塞甲醇水合物ⅱ的制备方法,包括以下步骤:将雷替曲塞溶解于水和甲醇溶剂的混合体系中,加热溶解,热抽,缓慢降温,加水析出,分离,干燥即得。其中,水和甲醇溶剂的体积比为1:1~1:6。
另一方面,本发明还提供了雷替曲塞乙醇水合物ⅲ的制备方法,包括以下步骤:将雷替曲塞溶解于水和乙醇溶剂的混合体系中,加热溶解,热抽,缓慢降温,加水析出,分离,干燥即得。其中,水和乙醇溶剂的体积比为1:3~1:7。
由于雷替曲塞粉末难溶于水,一般情况下,在有机溶剂中加入水,会使粗品的溶解度降低,这也是对比文件cn201210260633.7、cn201110361869.5中均采用无水的醇类或醚类溶剂对雷替曲塞粗品进行溶解的原因,但是发明人惊奇的发现在醇类溶剂中加入一定比例的水后反而会显著提升雷替曲塞的溶解度,水与醇类溶剂的比例对是否能够得到固态的中间体也至关重要。
以下通过试验来验证水与醇的比例对雷替曲塞晶型的影响、以及对其溶解度的影响,该试验在除水与醇的比例有差异外,其它试验条件均相同的情况下开展,具体试验结果详见下表。
表1来自初步小规模结晶结果的概要
本发明还发现以雷替曲塞溶剂化物ⅱ、ⅲ作为中间体,制备雷替曲塞一水合物的方法,通过该方法基本可以制备纯的雷替曲塞一水合物,且通过该方法制备的一水合物在长期稳定试验中,表现出了更稳定的特性。
以下是小规模稳定性试验的结果,雷替曲塞粗品可采用以n-[5-(n-甲胺基)-2-噻吩甲酰基]-l-谷氨酸二乙酯和2-甲基-6-溴甲基-3-氢-喹唑啉-4-酮为原料,经缩合、水解制备得雷替曲塞粗品或市场处购买雷替曲塞粗品,纯度约95%,水分3%。
批次1:按照us4992550(简称‘550)中公开的方法进行制备雷替曲塞一水合物;
批次2:将雷替曲塞粗品,重结晶生成雷替曲塞甲醇水合物ⅱ,在40-60℃下真空干燥6-8h,得雷替曲塞一水合物。
批次3:将雷替曲塞粗品,重结晶生成雷替曲塞乙醇水合物ⅲ,在50-65℃下真空干燥6-8h,得雷替曲塞一水合物。
以下对‘550方法和本申请的方法进行了比较分析,其具体区别详见下表
表2精制方法比较分析
从上述表中可以看出,本精制方法与‘550中公开的方法相比直接避免了nacl产生,而氯离子超标是该品种生产质量控制的难点。因此本发明操作简单,更加适合工业化操作。
与此同时,按照本申请制备的方法与‘550中公开制备方法相比,得到的成品的稳定性更高,以下通过稳定性试验来验证上述结论。
将上述3个批次的产品,在恒温恒湿的箱中进行30天的稳定性考察,试验条件:60℃/92.5%相对湿度(rh),在光照4000lx的条件下,放置10天,分别于0天,5天,10天,取样,进行产品纯度以及杂质检验。
表3批次1-3的稳定性试验的结果
由以上数据可知,以溶剂化物为中间体,直接干燥得精制品制备的雷替曲塞一水合物,在长期稳定性中更胜一筹。
附图说明
图1雷替曲塞甲醇水合物ⅱ的xrpd图谱;
图2雷替曲塞甲醇水合物ⅱ的dsc图谱;
图3雷替曲塞甲醇水合物ⅱ的tga图谱;
图4雷替曲塞甲醇水合物ⅱ的单晶空间结构图;
图5雷替曲塞乙醇水合物ⅲ的xrpd图谱;
图6雷替曲塞乙醇水合物ⅲ的dsc图谱;
图7雷替曲塞乙醇水合物ⅲ的tga图谱;
图8雷替曲塞无定型的xrpd图谱;
图9实施例9制备的雷替曲塞一水合物的hplc图谱;
图10实施例10制备的雷替曲塞一水合物的hplc图谱。
具体实施方式
下面结合具体实施例进一步说明本发明的技术方案,但并不限定本发明。
雷替曲塞粗品可采用以n-[5-(n-甲胺基)-2-噻吩甲酰基]-l-谷氨酸二乙酯和2-甲基-6-溴甲基-3-氢-喹唑啉-4-酮为原料,经缩合、水解制备得雷替曲塞粗品或市场处购买雷替曲塞粗品,纯度约95.0%,水分3%。
本发明检测药物晶型结构及其性能的仪器如下:
1.单晶结构由x射线单晶衍射仪测定,制造商荷兰enrafnoius&enrafnoius公司,仪器型号:cad4/pc。
2.粉末衍射仪是由制造厂商瑞士arl公司生产的,仪器型号:x'tra,cu-kα
3.差示扫描量热曲线和热重曲线由美国perkinelmer公司生产,仪器型号:pyris1dsc,采用氮气气氛,升温速率10℃/min。
4.卡尔费休水分测试仪型号:mettlertoledov30volumetrickftitrator
实施例1制备雷替曲塞甲醇一水合物ⅱ的粉末
将1g雷替曲塞溶解于40ml甲醇与10ml的水的混合溶剂中,加热至50℃溶清,热抽,自然缓慢降温至35℃,有晶体析出,缓慢滴加水110ml,低温5℃析晶2h,过滤干燥即得晶型ⅱ的白绿色晶体粉末。用xrpd、dsc、tg对其进行表征,结果如图1~3。
实施例2制备雷替曲塞甲醇一水合物ⅱ的单晶
将1g雷替曲塞粉末溶于水:甲醇(体积比=1:3)的混合溶剂中,通过0.45μm的滤纸将未溶解的固体过滤掉,将得到的饱和滤液用扎孔的薄膜封住,缓慢挥发至固体析出,过滤,真空干燥,收集固体即得晶型ⅱ。对单个晶体进行x射线单晶衍射,结果如图4。
实施例3制备雷替曲塞乙醇一水合物ⅲ的粉末
将1g雷替曲塞溶解于60ml乙醇与10ml的水的混合溶剂中,加热至50℃溶清,热抽,自然缓慢降温至40℃,加入雷替曲塞乙醇一水合物ⅲ晶种,有晶体析出,自然降温至35℃,缓慢滴加水170ml,低温5℃析晶2h,过滤干燥即得晶型ⅲ的晶体粉末。
实施例4纯有机溶剂中制备无定形
将1g雷替曲塞溶解于40ml甲醇中,加热至50℃溶清,热抽,自然缓慢降温至35℃,有晶体析出,缓慢滴加水110ml,低温5℃析晶2h,过滤干燥即得黄绿粉末,其xrd粉末衍射如图8所示,为无定形。
实施例5按照cn201110001785.0中公开的方法进行制备
将0.20g(0.38mmol)n-[5-[n-[(3,4-二氢-2-甲基-4-氧代-6-喹唑啉基)甲基]-n-甲基氨基]-2-噻吩甲酰]-l-谷氨酸二乙酯溶于10ml乙醇和10ml丙酮中,加入3mol·l-1氢氧化钠水溶液14ml,室温搅拌反应3h。减压蒸馏蒸除乙醇和丙酮,用2mol·l-1盐酸调ph=4,过滤,得棕黄色固体,用甲醇-无水乙醚重结晶,得浅黄色固体雷替曲塞0.14g,收率80.1%。经检测其为无定形粉末。
实施例6按照cn201210260633.7中公开的方法进行制备
温度为0℃,往480ml1nnaoh溶液中加入分次加入52.8gn-[5-[n-甲基-n-(2-甲基-4-氧代-3,4-二氢喹唑啉基-6-甲基)氨基]噻吩-2-甲酰基]-l-谷氨酸二乙酯(约0.1mol)纯度在97%,继续搅拌2小时,过滤。滤液用二氯甲烷(500ml×2)萃取掉有机相杂质,再用乙酸乙酯(500ml×2)再萃取掉剩余的有机相杂质,在搅拌下(0--5℃)水层用1n盐酸调ph值至3.0左右,有大量固体析出。0℃下搅拌1小时,过滤,产物用纯化水洗涤,洗至滤液无cl-离子残留用0.1nagno3测定),尽量抽干,得雷替曲塞粗湿品138克,取该雷替曲塞粗产物13.8g加入到600ml无水甲醇中,室温搅拌0.5小时,试制品有少量不溶物,室温下过滤,滤液28-35℃下浓缩析晶,置4℃冰箱中冷藏3小时,过滤,产物用少量纯化水洗涤,室温真空干燥10小时,得几近白色的结晶固体3.2g,经检测其为无定型粉末。
实施例7按照cn201410844732.9中公开的方法进行制备
往反应瓶中加入5g雷替曲塞粗品,再加入15ml甲醇,水浴加热,回流搅拌,使其全部溶解,回流0.5h,将溶液降温至40℃,于40℃保温静置1h,过滤去掉杂质,再将溶液在4小时内缓慢降温至15℃,保温7h,固体析出。经检测其为无定形粉末。
实施例8按照cn20110361869.5中公开的方法进行制备
粗品置于1000ml反应瓶中,加入600ml甲醇加热回流至澄清,过滤,滤液冷却至5-10℃,慢慢加入约4200ml纯化水搅拌析晶,过滤,固体用纯化水洗涤,得淡黄色固体。40℃真空干燥24小时,得浅黄色精品30.3g,摩尔收率85%。hplc纯度99.8%。经检测其为无定形粉末。
实施例9对无定型、一水合物、溶剂化合物进行长期稳定试验考察。
将雷替曲塞无定型、一水合物、溶剂化物ⅱ、ⅲ在恒温恒湿箱中进行6个月的稳定性试验。试验条件为:25℃/75%相对湿度(rh),分别于0、1、2、3、6个月取样,进行纯度和杂质检验。
表4不同晶型稳定性试验结果
从上表数据可知,雷替曲塞的甲醇水合物以及乙醇水合物在长期稳定性试验中,表现出了比无定型更高的稳定性,并且其稳定性并不劣于一水合物的稳定性。
实施例10
取实施例1所得雷替曲塞甲醇水合物2g,在40-60℃下真空干燥6-8小时后即得雷替曲塞一水合物,收率为86%,其纯度为99.94%,最大单杂0.04%,其hplc图谱详见图9。
实施例11
取实施例3所得雷替曲塞乙醇水合物3g,在50-65℃下真空干燥6-8h后,即得雷替曲塞一水合物,收率为83%,纯度为99.84%,最大单杂为0.06%,其hplc图谱详见图10。
- 还没有人留言评论。精彩留言会获得点赞!