索法地尔杂质B及其制备方法和应用与流程
本发明涉及一种治疗和预防中枢神经系统的急慢性神经变性疾病药物索法地尔的杂质及其制备方法,尤其涉及一种索法地尔杂质B及其制备方法和应用,属于医药化工领域。
背景技术:
索法地尔(Salfaprodil),化学名为2-羟基-5-{[2,3,5,6-四氟-4-(三氟甲基)苄基]氨基}苯甲酸钾,其结构式如下所示:
索法地尔是一种中等强度的N-甲基-D-天冬氨酸(NMDA)受体拮抗剂,也是一种强力的抗氧化剂,具备双重神经保护作用,可同时阻断NMDA受体介导的兴奋毒性和机体的氧化应激反应,其保护神经功能的效果可持续超过28天,提供了广泛的神经保护作用,扩大了治疗时间窗。
公告号为CN1309703C的中国专利申请公开了索法地尔可用于治疗脑血管和神经系统疾病和症状中的常规或病理性神经性疾病。具体而言,索法地尔用于预防和治疗血栓栓塞、缺血性中风、出血性中风、脑血管痉挛、脑老化、外伤性脑损伤、外伤性脊髓损伤、心搏停止、动脉低血压、低血糖症、缺氧症和组织缺氧。索法地尔还可以有效地用于缓解神经变性疾病,如亨廷顿症、阿尔茨海默氏症、老年痴呆、小脑退化症、肌萎缩性脊髓侧索硬化、帕金森症、唐氏综合症、癫痫、多发梗塞性痴呆和脑炎。
药物中所含有的杂质一般会降低药物的疗效,甚至在有些情况下会产生严重的毒副作用,随着人们的健康意识越来越高,对药物中的杂质含量的要求越来越高。因此,确定索法地尔药物中未知杂质的准确结构具有重要的意义,一方面可以对杂质含量进行更有效的控制,另一方面可以确定杂质的毒副作用。
技术实现要素:
本发明提供了一种新的化合物索法地尔杂质B及其制备方法和应用,该化合物可以用于索法地尔质量分析时的对照品。
一种索法地尔杂质B,其结构如式(Ⅰ)所示:
所述索法地尔杂质B的分子式为C15H9F6NO3。
试验结果表明,式(I)所示的新化合物为现有的索法地尔生产过程中出现的一项杂质(记为索法地尔杂质B),该杂质含量大小关系到索法地尔的成品的质量。本发明确定了该杂质的准确结构,从而可以对索法地尔中该杂质的含量进行准确的定量,以帮助生产过程中对该杂质含量的控制。此外,如有必要还可以深入分析该杂质的存在可能对活性产生何种影响,提高索法地尔的用药安全性。
所述的索法地尔杂质B具有1HNMR(500MHz,DMSO)δ(ppm):7.72(t,1H,Ar),7.07(d,1H,Ar),6.93(dd,1H,Ar),6.76(d,1H,Ar),4.34(s,2H,CH2),其中各峰±0.1ppm。
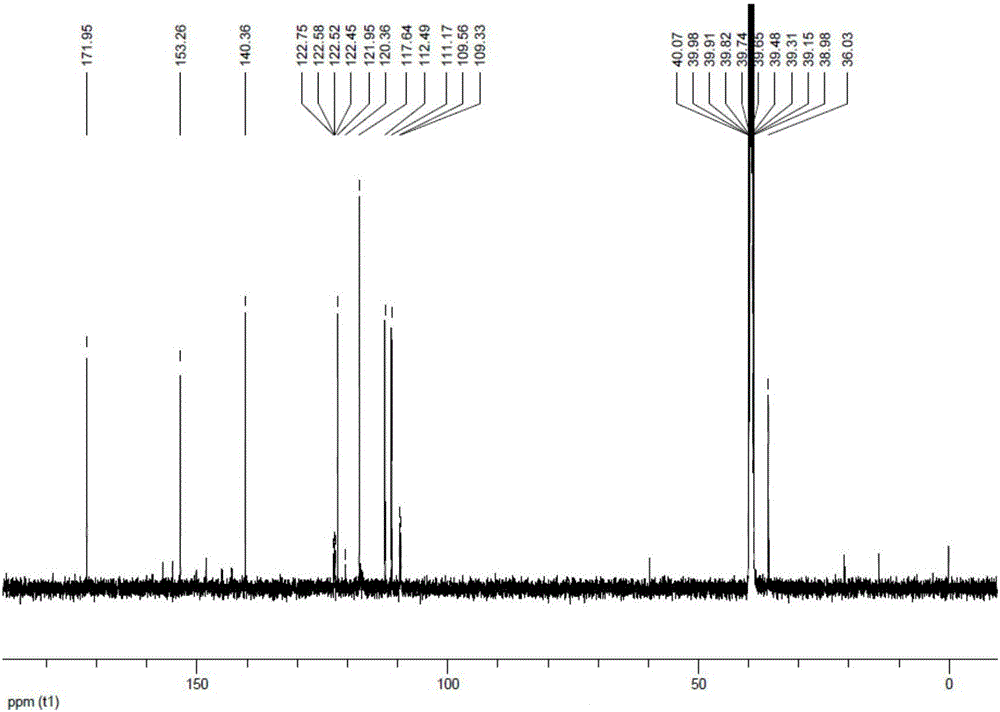
所述索法地尔杂质B具有13CNMR(125MHz,DMSO)δ(ppm):171.95,153.26,140.36,122.52,120.36,117.64,112.49 111.17,109.45,36.03,其中各峰±0.1ppm。
本发明还提供了一种索法地尔杂质B的制备方法,以索法地尔为起始原料,经酸化,还原反应得到该杂质B,具体包括以下步骤:
步骤A:化合物(Ⅱ)在酸存在下经酸化得到化合物(Ⅲ),反应式如下:
步骤B:化合物(Ⅲ)在碱和催化剂存在下经还原得到化合物(Ⅰ),反应式如下:
步骤A操作过程如下:化合物(Ⅱ)溶于水中,加酸调节至pH 2~4,萃取,得到化合物(Ⅲ)溶液,直接进入步骤B进行反应。
进一步的,步骤A中,化合物(Ⅲ)溶液可进行适当浓缩。
步骤A中,所述的酸为盐酸、硫酸、磷酸、氢溴酸、硝酸、草酸、甲酸中的至少一种,优选为盐酸。
步骤A中,萃取所用溶剂为C1-C6脂肪醇与C1-C6脂肪酸形成的酯(例如醋酸异丙脂或乙酸乙酯)、C6-C10醚类中的至少一种。
步骤A中,浓缩的化合物(Ⅲ)溶液可直接用于下一步反应;也可将化合物(Ⅲ)溶液浓缩至干,纯化后进行下一步反应。
步骤B操作过程如下:将化合物(Ⅲ)溶液、催化剂与碱混合,在氢气存在下进行还原反应,反应结束后,反应液经过滤,浓缩滤液,浓缩液用水溶解,调pH至酸性(pH2~4),萃取,浓缩得到粗产品,再经过柱层析精制得到杂质B。
步骤B中,以纯化的化合物(Ⅲ)为原料时,需加入溶剂进行反应,所述溶剂为步骤A萃取选用的溶剂。
步骤B中,所述催化剂为:1%-30%的钯碳,钯黑,以及其它载体负载的钯,醋酸钯,氢氧化钯,氯化钯等钯盐,镍,铂等中的一种或其组合物;优选5%-20%的钯碳,雷尼镍。
步骤B中,所述碱为无机碱或有机碱,所述无机碱为锂、钠、钾相应的氢氧化物、碳酸盐、碳酸氢盐、氨水中的至少一种;
所述的有机碱为C1-C6脂肪酸的锂、钠、钾盐,三乙胺,二乙胺,吡啶,2-甲基吡啶,4-二甲氨基吡啶,N-甲基吗啉,四甲基乙二胺中的至少一种。
步骤B中,反应溶剂为C1-C6脂肪醇以及其与C1-C6脂肪酸形成的酯、C6-C10脂肪烃、醚类中的一种或两种及以上的混合物。
步骤B后处理中,萃取所用溶剂为C1-C6脂肪醇与C1-C6脂肪酸形成的酯,C6-C10醚类中的至少一种。
步骤B中,反应温度为10℃~200℃,反应压力为常压~10Mpa。
步骤B中,打浆精制所用的溶剂为C6-C10脂肪烃、卤代烃、芳香烃或取代芳香烃。
本发明所述索法地尔杂质B的制备方法,反应原料的用量并没有严格的限定,一般按照化学反应计量比进行反应,也可过量进行反应;各步的反应溶剂和反应试剂的用量并没有严格的限定,可根据反应原料的用量调整:反应原料较多增加反应溶剂和反应试剂的用量,反应原料较少减少反应溶剂和反应试剂的用量;各步骤的反应溶剂可根据本领域技术人员的只是进行选择,如醇类、酮类、醚类等;各步骤的后处理方法可根据本领域技术人员的知识进行选择,如萃取,蒸馏等。
本发明还提供了一种所述的索法地尔杂质B在索法地尔的质量控制中的应用,所述的索法地尔杂质B作为对照品。采用该索法地尔杂质B作为对照品,可以对索法地尔中的杂质进行定性和定量分析,该定性和定量分析方法为HPLC法。
同现有技术相比,本发明的有益效果体现在:
本发明合成高纯度的索法地尔杂质B,可作为索法地尔成品检测分析中的杂质B标准品,从而提升索法地尔成品检测分析对杂质B的准确定位和定性,有利于加强对该杂质的控制,进而提高索法地尔成品质量;同时,可以研究该杂质对药物活性和毒副作用的影响。
附图说明
图1为索法地尔杂质B的核磁共振氢谱谱图;
图2为索法地尔杂质B的核磁共振碳谱谱图;
图3为按照CN1309703C的方法得到的索法地尔的HPLC谱图;
图4为实施例4索法地尔杂质B的HPLC谱图。
具体实施方式
以下结合具体实施例对本发明作进一步详细说明
实施例1化合物(Ⅲ)的合成
将42.13g化合物(Ⅱ)溶于210mL水中,降温至0~10℃,向其中加入盐酸,调节pH=2~4,室温搅拌半个小时。用乙酸乙酯萃取三次,合并有机相,并用饱和食盐水洗涤一次,无水硫酸钠干燥;过滤除去硫酸钠,滤液减压浓缩得到化合物(Ⅲ)38.1g,收率99%,HPLC纯度100%。反应式如下:
实施例2化合物(Ⅲ)的合成
将42.13g化合物(Ⅱ)溶于210mL水中,降温至0~10℃,向其中加入盐酸,调节pH=2~4,室温搅拌半个小时。用醋酸异丙脂萃取三次,合并有机相,并用饱和食盐水洗涤一次,无水硫酸钠干燥;过滤除去硫酸钠,滤液减压浓缩得到化合物(Ⅲ)37.2g,收率97%,HPLC纯度100%。
实施例3化合物(Ⅰ)的合成
将38.1g实施例1制备的化合物(Ⅲ)溶于250mL甲醇中,向其中加入3.8g含量为5%的钯碳,13.8g无水碳酸钾;反应釜先用氮气置换三次,再用氢气置换三次后,控制压力1.0~2.0Mpa,温度40~50℃反应。反应8h后,取样进行HPLC检测,原料含量小于等于5%,停止反应。反应液过滤除去钯碳,滤液减压浓缩除去甲醇。向浓缩液中加入100mL水,并用盐酸调节pH2~4。用乙酸乙酯萃取三次,合并有机相,并用饱和食盐水洗涤一次,无水硫酸钠干燥;过滤除去硫酸钠,滤液减压浓缩得到粗品化合物(Ⅰ)23.4g,粗品收率64%。
化合物(Ⅰ)粗品23.4g,经硅胶柱层析纯化。再经120mL正己烷室温打浆5h,过滤,滤饼用冷的正己烷洗涤两次得到化合物(Ⅰ)17.8g,即为索法地尔杂质B,收率76%,HPLC纯度99%。
1HNMR(500MHz,DMSO)δ(ppm):7.72(t,1H,Ar),7.07(d,1H,Ar),6.93(d,1H,Ar),6.76(d,1H,Ar),4.34(s,2H,CH2)。
13CNMR(125MHz,DMSO)δ(ppm):171.95,153.26,140.36,122.52,120.36,117.64,112.49,111.17,109.45,36.03。
反应式如下:
实施例4化合物(Ⅰ)的合成
将38.1g实施例1制备的化合物(Ⅲ)溶于250mL乙醇中,向其中加入7.6g活化的雷尼镍,15.1g三乙胺;反应釜先用氮气置换三次,再用氢气置换三次后,控制压力2.5Mpa,温度60℃反应。反应20h后,取样进行HPLC检测,原料含量小于等于5%,停止反应。反应液过滤除去催化剂,滤液减压浓缩除去乙醇。向浓缩液中加入100mL水,并用盐酸调节pH 2~4。用乙酸乙酯萃取三次,合并有机相,并用饱和食盐水洗涤一次,无水硫酸钠干燥;过滤除去硫酸钠,滤液减压浓缩得到粗品化合物(Ⅰ)19.0g,粗品收率52%。
化合物(Ⅰ)粗品19.0g,经硅胶柱层析纯化。再经95mL正庚烷室温打浆5h,过滤,滤饼用冷的正庚烷洗涤两次得到化合物(Ⅰ)13.5g,收率71%,色谱纯度99%。
实施例5化合物(Ⅰ)的合成
将38.1g实施例1制备的化合物(Ⅲ)溶于250mL甲醇中,向其中加入3.8g含量为10%的钯碳,13.8g无水碳酸钾;反应釜先用氮气置换三次,再用氢气置换三次后,控制压力1.0~2.0Mpa,温度40~50℃反应。反应8h后,取样进行HPLC检测,原料含量小于等于5%,停止反应。反应液过滤除去钯碳,滤液减压浓缩除去甲醇。向浓缩液中加入100mL水,并用盐酸调节pH2~4。用乙酸乙酯萃取三次,合并有机相,并用饱和食盐水洗涤一次,无水硫酸钠干燥;过滤除去硫酸钠,滤液减压浓缩得到粗品化合物(Ⅰ)24.5g,粗品收率67%。
化合物(Ⅰ)粗品24.5g,经硅胶柱层析纯化。再经120mL甲苯室温打浆5h,过滤,滤饼用冷的甲苯洗涤两次得到化合物(Ⅰ)18.4g,收率75%,HPLC纯度99%。
实施例6索法地尔杂质B的定性分析
参照专利CN1309703C的合成方法制备的索法地尔样品,采用高效液相色谱法测得该索法地尔样品的图谱,见附图3。
在完全相同的条件下,采用高效液相色谱法测得实施例3制备的索法地尔杂质B(Ⅰ)的图谱,见附图4。
图3和图4对比可知,实施例3制备的索法地尔杂质B的出峰位置为8.5min,与索法地尔样品中的8.5min处杂质出峰时间的误差在2s以内,两者为同一种物质。
- 还没有人留言评论。精彩留言会获得点赞!