用于生产无包膜病毒颗粒的方法与流程
本发明涉及以高纯度且不经过辛苦操作生产无包膜病毒颗粒的方法。
背景技术:
在基因重组领域或医学领域中,对于将基因引入包括人的哺乳动物的细胞中,目前已经使用了使用电穿孔或金属微粒的物理方法、使用核酸、聚阳离子或脂质体的化学方法、以及使用病毒衍生的用于基因转移的载体(在下文中,被称作病毒载体)的生物学方法。病毒载体意指通过改变天然病毒使得所述病毒可以将期望的基因等转移进靶标中而得到的载体,并且这样的载体的开发最近已经进步。通过基因重组技术制备的病毒载体通常被称作重组病毒载体。从其中衍生出重组病毒载体的病毒的众所周知的例子包括带有包膜的病毒例如逆转录病毒、慢病毒、仙台病毒和疱疹病毒,和不带有包膜的病毒(无包膜病毒)例如腺病毒和腺伴随病毒(在下文中,被称作aav)。
具体地,aav可以感染包括人细胞的多种细胞,并且aav甚至感染其中分化终止的非分裂细胞,包括血细胞、肌细胞和神经细胞。另外,由于aav对人不是病原性的,它具有低副作用风险。aav的病毒颗粒是物理化学上稳定的。因为这些原因,对于作为在基因治疗中应用的用于基因转移的载体的实用价值,aav最近已经引起注意,所述基因治疗用于治疗先天性遗传疾病以及治疗癌症或感染。
生产重组病毒载体的方法经常包括,将病毒颗粒形成所必需的元件以一个或多个核酸构建体的形式引入细胞中以产生具有生产病毒的能力的细胞(在下文中,被称作病毒生产细胞),和培养所述细胞以表达所述病毒颗粒形成所必需的元件。一般而言,在所述病毒颗粒形成所必需的元件中,将需要以顺式提供的元件和可以以反式提供的元件分别地引入用于病毒生产的细胞,由此防止野生型病毒的生产和重组病毒在被该病毒感染的宿主中的自我复制(专利文献1)。
在下文中,作为一个例子,具体地解释从aav衍生的重组载体(在下文中,被称作raav)。一种确立的方法包括:1)引入raav质粒,其中保留放置在野生型aav基因组的每个末端处的itr,且除去rep和cap基因,和2)引入用于表达rep和cap基因的质粒以反式提供rep和cap蛋白,和3)用腺病毒感染,因为aav需要提供来自被称为辅助病毒的任意病毒诸如腺病毒、疱疹病毒或痘苗病毒的补充元件用于形成感染性病毒颗粒。通过上述方法得到的载体溶液在理论上被腺病毒污染。为了避免腺病毒污染,已经开发了一种生产载体的方法,其替代上述3)包括3')引入辅助质粒,在腺病毒衍生的元件中,其仅表达aav病毒颗粒的形成所必需的元件(无辅助系统)。
将已经完成病毒生产的病毒生产细胞收集和均质化以得到含有raav颗粒的细胞匀浆。对细胞匀浆进行合适的步骤诸如用滤器过滤、超速离心、层析或超滤,以将raav颗粒纯化为终产物。
目前,由于病毒载体的用途被延伸至用于基因治疗的基础研究或临床应用的领域,需要以较高滴度和较高纯度得到病毒颗粒的方法。公开了多种改善的方法。例如,已知一种增加病毒颗粒生产和所述病毒颗粒向培养上清中的释放速率的方法,其包括,在其中培养基具有升高的ph的胁迫条件下培养病毒生产细胞(专利文献1)。其它方法包括对生产的病毒的纯化及其以后的步骤的改善。对于raav颗粒的纯化,建议了不应用超速离心或层析的纯化各种血清型的raav颗粒的方法(非专利文献1和非专利文献2)。但是,所述纯化方法包括4个步骤:聚乙二醇(peg)相和水相的双相分离,用peg沉淀,用氯仿处理,和透析;并且因此是复杂的。此外,通过该纯化方法纯化的产物包含sds-page上的许多条带,其可能来自杂质。因此,该纯化方法从纯度和简便性的角度而言不是足够的。因此,在raav颗粒纯化之前执行的病毒生产细胞的处理中仍然存在改善的空间。
引文列表
专利文献
专利文献1:wo2000/14205
非专利文献
非专利文献1:j.virol.methods,vol.183,no.2,pp.139-146,2012
非专利文献2:bioengineered,vol.4,no.2,pp.103-106,2013。
技术实现要素:
本发明要解决的问题
本发明的一个目的是,提供以高纯度和不经过辛苦操作生产无包膜病毒颗粒的方法。
问题的解决方案
针对提供以高纯度和不经过辛苦操作生产无包膜病毒颗粒的方法的目的,本发明人专心地进行了研究,结果发现,通过向包含无包膜病毒颗粒的中性或碱性样品加入降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质,和除去在酸化加入该物质后的样品的步骤中形成的沉淀物,获得包含无包膜病毒颗粒的级分。因而,完成了本发明。
本发明提供:
[1]生产无包膜病毒颗粒的方法,所述方法包括:
(a)向包含无包膜病毒颗粒的中性或碱性样品加入降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质的步骤,
(b)酸化加入了该物质的样品的步骤,
(c)除去步骤(b)中形成的沉淀物以获得包含无包膜病毒颗粒的级分的步骤;
[2]根据[1]的方法,其在步骤(c)之后进一步包括(d)纯化无包膜病毒的步骤;
[3]根据[1]或[2]的方法,其中降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质是表面活性剂;
[4]根据[1]至[3]中任一项的方法,其中酸化步骤是加入酸性溶液的步骤;
[5]根据[1]至[4]中任一项的方法,其中包含无包膜病毒颗粒的中性或碱性样品是无包膜病毒生产细胞的溶解物或匀浆、含有无包膜病毒颗粒的无包膜病毒生产细胞的培养上清、或从无包膜病毒生产细胞获得的提取物;
[6]根据[5]的方法,其中包含无包膜病毒颗粒的中性或碱性样品是通过用酸性溶液处理无包膜病毒生产细胞获得的提取物的中和产物;
[7]根据[1]至[6]中任一项的方法,其中包含无包膜病毒颗粒的中性或碱性样品是用核酸酶处理的样品;
[8]根据[1]至[7]中任一项的方法,其中无包膜病毒颗粒是腺伴随病毒颗粒;
[9]试剂盒,其用于实施根据[1]至[8]中任一项的方法,所述试剂盒包括:
(1)降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质,和
(2)酸性溶液;和
[10]根据[9]的试剂盒,其进一步包括中和溶液。
发明效果
根据本发明,提供以高纯度和不经过辛苦操作生产无包膜病毒颗粒的方法。
附图说明
[图1]图1显示实施例1中制备的样品的sds-page的结果。
[图2]图2显示实施例1中制备的样品的回收率。
[图3]图3显示实施例2中制备的样品的sds-page的结果。
[图4]图4显示实施例3中制备的样品的sds-page的结果。
[图5]图5显示实施例6中制备的样品的sds-page的结果。
具体实施方式
本文中使用的无包膜病毒指的是除了有包膜病毒以外的病毒。有包膜病毒指的是在病毒表面上具有脂质层或脂质双层的病毒。无包膜病毒的代表性例子包括dna基因组病毒,例如腺病毒、细小病毒、乳多空病毒和人乳头瘤病毒,和rna基因组病毒,例如轮状病毒、柯萨奇病毒、肠道病毒、扎幌病毒、诺如病毒、脊髓灰质炎病毒、埃可病毒、甲型肝炎病毒、戊型肝炎病毒、鼻病毒和星状病毒。
通过本发明的生产方法生产的无包膜病毒包括但不限于,已知其生产方法的无包膜病毒,从自然界新得到的无包膜病毒,和从上述无包膜病毒衍生的基因重组病毒载体。通过本发明的生产方法生产的无包膜病毒的优选例子包括腺病毒以及属于细小病毒科的aav。本发明的生产方法适于任何已知血清型的aav的生产。例如,本发明的生产方法可用于选自aav1、aav2、aav3、aav4、aav5、aav6、aav7、aav8、aav9、aav10、aav11、aav12和aav13的至少一种aav的生产。如本文所用的,raav的血清型基于raav的衣壳由其衍生的aav的血清型。换言之,raav的血清型取决于用于制备raav的cap基因的来源而确定,并且不取决于raav颗粒中包封的aav基因组的血清型。例如,当raav的衣壳衍生自aav6且raav颗粒中包封的aav基因组中的itrs衍生自aav2时,如本文所用的,raav的血清型是6。此外,本发明的生产方法适于包含上述血清型的aav衣壳的变体的aav的生产。
如本文所用的,"病毒颗粒"意味着由称为衣壳的蛋白壳构成的颗粒。如本文所用的,"病毒载体"意味着病毒颗粒中包含的病毒基因组(核酸形式)。例如,在aav的情况下,raav颗粒是由称为衣壳的蛋白壳构成的颗粒并且包括raav载体。raav载体包含raav颗粒中存在的病毒基因组dna。如本文所用的,"病毒颗粒"包括不包含病毒基因组的病毒样颗粒(例如,aav中空颗粒:wo2012/144446)。病毒颗粒包括但不限于,衍生自raav载体的病毒样颗粒和aav中空颗粒。
如本文所用的,"酸性"意味着处于低于ph6.5的ph,"中性"意味着处于ph6.5-7.5,并且"碱性"意味着处于超过ph7.5的ph。
下文对本发明进行解释。
(1)本发明的无包膜病毒颗粒的生产方法
本发明的无包膜病毒颗粒的生产方法包括:
(a)向包含无包膜病毒颗粒的中性或碱性样品加入降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质的步骤,
(b)酸化加入了该物质的样品的步骤,
(c)除去步骤(b)中形成的沉淀物以获得包含无包膜病毒颗粒的级分的步骤。
具体地,首先,将降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质加入包含无包膜病毒颗粒的中性或碱性样品。该物质的加入可通过加入固体物质实施,或优选地通过以期望的最终浓度加入预先制备的溶液实施。样品可在该物质的加入之后立即接受下一步骤(b),或优选地,样品在该物质的加入之后静置一会儿。样品静置的温度和样品静置的时间可以适当确定,并且没有特别限定。例如,样品静置的温度是0至40℃,优选地4至37℃。例如,样品静置的时间是1分钟至24小时,优选地5分钟至1小时。例如,样品在37℃静置30分钟。
本发明方法中应用的"降低在酸性条件下的蛋白的溶解度的物质"意味着,与中性或碱性条件相比,具有降低在酸性条件下的蛋白的溶解度的特性的物质。该物质没有特别限定,并且优选地是具有与样品中的蛋白相互作用以降低在酸性条件下的蛋白的溶解度并使蛋白沉淀的特性的物质。
本发明方法中应用的"在酸性条件下沉淀的物质"意味着,具有通过从中性或碱性条件变为酸性条件而自身沉淀的特性的物质。该物质没有特别限定,并且优选地是具有在酸性条件下自身与样品中的蛋白一起沉淀的特性的物质。
本发明方法中应用的降低在酸性条件下的蛋白的溶解度的物质和在酸性条件下沉淀的物质可以是不同的物质或可以是相同的物质。降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质的例子包括但不限于表面活性剂。该表面活性剂被大致分类为阴离子表面活性剂、非离子表面活性剂、阳离子表面活性剂和兼性表面活性剂。
阴离子(anionic)表面活性剂也称为阴离子(anion)表面活性剂,并且在水中电离成为有机阴离子。阴离子表面活性剂分类为羧酸型、磺酸型、硫酸酯型和磷酸酯型。羧酸型的例子包括脱氧胆酸盐、胆酸盐和月桂酰肌氨酸盐。磺酸型的例子包括十二烷基硫酸盐。盐的优选例子包括钠盐和锂盐。
非离子表面活性剂在水中不电离。非离子表面活性剂分类为酯型、醚型、酯醚型、链烷醇酰胺型、糖型和甲葡糖胺型。酯型的例子包括聚氧乙烯失水山梨醇单月桂酸酯[tween(注册商标)20]、聚氧乙烯失水山梨醇单棕榈酸酯[tween(注册商标)40]、聚氧乙烯失水山梨醇单硬脂酸酯[tween(注册商标)60]和聚氧乙烯失水山梨醇单油酸酯[tween(注册商标)80]。醚型的例子包括聚氧乙烯壬基酚醚[nonidet(注册商标)p-40]和聚氧乙烯烷基醚[brij(注册商标)l23,brij(注册商标)l58]。酯醚型的例子包括聚氧乙烯失水山梨醇脂肪酸酯、聚氧乙烯己糖醇酐脂肪酸酯和失水山梨醇脂肪酸酯聚乙二醇。链烷醇酰胺的例子包括n,n-二[3-(d-葡萄糖酰氨基)丙基]脱氧胆酰胺。糖型的例子包括烷基麦芽糖苷(alkylmaltoside)和烷基吡喃葡萄糖苷(alkylglucopyranoside)。甲葡糖胺型的例子包括烷基-n-甲葡糖胺。
阳离子(cationic)表面活性剂也称为阳离子(cation)表面活性剂,并且在水中电离成为有机阳离子。阳离子表面活性剂分类为烷基胺盐型和季铵盐型。烷基胺盐型的例子包括一甲胺盐酸盐、二甲胺盐酸盐和三甲胺盐酸盐。季铵盐型的例子包括溴化十六烷基三甲铵和溴化肉豆蔻基三甲铵。
兼性表面活性剂意味着分子内既具有阴离子基团又具有阳离子基团的表面活性剂。在水溶液中电离的兼性表面活性剂与氨基酸类似,并且兼性表面活性剂包括许多氨基酸衍生物。因此,兼性表面活性剂具有类似氨基酸的等电点。当兼性表面活性剂在比等电点更碱性的条件下存在时,其充当阴离子表面活性剂,而当兼性表面活性剂在比等电点更酸性的条件下存在时,其充当阳离子表面活性剂。兼性表面活性剂的例子包括但不限于,磺基甜菜碱如月桂基磺基甜菜碱、辛酰基磺基甜菜碱、辛基磺基甜菜碱、棕榈基磺基甜菜碱和肉豆蔻基磺基甜菜碱。
在本发明方法中,优选应用在酸性ph不溶解的阴离子表面活性剂。更优选地,应用羧酸型阴离子表面活性剂如脱氧胆酸钠、鹅脱氧胆酸钠、胆酸钠、甘氨胆酸钠、牛磺胆酸钠或牛磺脱氧胆酸钠。尤其优选地,应用脱氧胆酸钠、鹅脱氧胆酸钠或胆酸钠。
在本发明方法步骤(a)中,降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质可以作为固体物质加入,或可以作为预先制备的物质溶液加入。溶液中物质的浓度没有特别限定,并且可以由本领域技术人员适当确定。用于制备物质溶液的溶剂没有特别限定,并且可以适当选自水、缓冲液、用于细胞培养的培养基等。将降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质加入包含无包膜病毒颗粒的中性或碱性样品使得物质的浓度成为期望的最终浓度。物质的期望的最终浓度没有特别限定,只要它是这样的浓度而允许样品中的污染蛋白在酸性条件下沉淀。虽然物质的期望的最终浓度取决于样品中包含的无包膜病毒的种类或物质的种类,例如,当应用脱氧胆酸钠、鹅脱氧胆酸钠或胆酸钠时,最终浓度是例如0.1至2%(v/v),优选地0.2至1%(v/v)。当降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质的最适浓度取决于无包膜病毒的种类变化时,确定物质的适当浓度是自然的。
然后,在本发明方法中,其中加入物质的样品的ph被酸化。这一步骤可以通过任何方法实施,只要该方法能够酸化中性或碱性样品,并且该方法没有特别限定。用于酸化样品的手段可以是样品中气态酸的鼓泡或固体酸的加入。尤其优选地,加入预先制备的酸性溶液,使得成为期望的最终浓度。本发明中应用的酸的例子包括选自柠檬酸、乙酸、苹果酸、磷酸、盐酸、硫酸、硝酸、乳酸、丙酸、丁酸、草酸、丙二酸、琥珀酸、富马酸、马来酸、酒石酸、苯甲酸、磺基水杨酸、甲酸及它们的盐、以及具有低于ph6.5的缓冲区的good氏缓冲液如mes和bis-tris的化合物。作为酸性溶液,例如,应用含有上述化合物的溶液。用于制备酸性溶液的溶剂没有特别限定。溶剂可以适当选自水、缓冲液、用于细胞培养的培养基等。尤其优选地,应用包含柠檬酸或其盐的水溶液。将酸或酸性溶液加入样品使得成为期望的最终浓度。期望的最终浓度没有特别限定,只要它是这样的浓度而允许样品的ph成为酸性。酸化样品之后,样品的最终ph优选地低于ph6.5,更优选地在ph4至6的范围。当样品的最适ph取决于无包膜病毒的种类变化时,确定样品的适当ph是自然的。
本发明方法中应用的包含无包膜病毒颗粒的样品可以是衍生自无包膜病毒生产细胞的样品。该病毒生产细胞包括但不限于,得自环境或来自具有感染的患者的临床样品的病毒生产细胞,和人工制备的病毒生产细胞。
用于制备无包膜病毒生产细胞的细胞没有特别限定。用于制备无包膜病毒生产细胞的细胞的例子包括哺乳动物诸如人、猴和啮齿动物的细胞,且其优选例子包括具有高转化效率的细胞如293细胞(atcccrl-1573)、293t/17细胞(atcccrl-11268)、293f细胞、293ft细胞(都由lifetechnologiescorp.制造)、g3t-hi细胞(wo2006/035829)、sf9细胞(atcccrl-1711)和aav293细胞(由stratagenecorp.制造),aav293细胞为商购可得的用于病毒生产的细胞系。例如,293细胞等恒定地表达腺病毒e1蛋白。也可以使用这样的细胞:其被修饰成短暂地或恒定地表达raav生产所需的一种或一些蛋白。在这些各种细胞中,通过应用已知方法或商购可得的试剂盒可以引入无包膜病毒载体以获得无包膜病毒生产细胞。无包膜病毒生产细胞的培养可以在已知培养条件下实施。例如,细胞在30至37℃的温度、95%的湿度和5至10%(v/v)的co2浓度培养。但是,无包膜病毒生产细胞的培养条件不限于上述培养条件。所述细胞培养可以在上述范围以外的温度、湿度和co2浓度进行,只要完成期望的细胞生长和无包膜病毒的生产。培养期没有特别限制,并且例如,细胞培养持续12-150小时,优选48-120小时。
下文,作为无包膜病毒生产细胞的例子,对raav生产细胞进行解释。raav生产细胞可以如下生产:在任何细胞中引入:(a)衍生自aav的编码rep蛋白的核酸和编码cap蛋白的核酸,和(b)提供腺病毒衍生元件例如e1a蛋白、e1b蛋白、e2蛋白、e4蛋白和varna的核酸,作为对于raav颗粒的形成所必需的元件,和(c)要包封在raav颗粒中的核酸。其中替代上述(b)中描述的核酸应用腺病毒等作为辅助病毒的方法也是已知的。
衍生自aav的编码rep蛋白的核酸和编码cap蛋白的核酸、以及提供腺病毒衍生元件的核酸可以是任何形式,其没有限定。这些核酸可以作为一个或多个能够提供所述元件的核酸构建体插入质粒或病毒载体,然后该质粒或病毒载体可以引入细胞。这些核酸向细胞中的引入可以通过已知方法应用商购可得的或已知的质粒或病毒载体实施。
要包封在raav颗粒中的核酸由衍生自aav的itr序列和期望被raav载体负载的核酸组成。期望被raav载体负载的核酸的例子包括任何外来基因,例如用于提供多肽(酶、生长因子、细胞因子、受体、结构蛋白等)的核酸、反义rna、核酶、诱饵(decoy)、诱导rna干扰的rna等。另外,为了控制外来基因的表达,可以将合适的启动子、增强子、终止子和其它转录调节元件插入核酸中。例如,要包封在raav颗粒中的核酸可以含有期望被raav载体在2个itr序列之间负载的任何外来基因,或可以含有期望被raav载体负载的任何外来基因和至少一个元件,所述元件用于控制2个itr序列之间的外来基因的表达。要包封在病毒颗粒中的核酸可以作为核酸构建体以质粒的形式引入细胞中。例如,通过使用商购可得的paav-cmv载体(由takarabioinc.制造)等,可以构建质粒。
本发明方法中应用的包含无包膜病毒颗粒的中性或碱性样品可以通过例如如上述培养的无包膜病毒生产细胞的破坏或裂解而制备。在无包膜病毒生产细胞培养期间无包膜病毒颗粒泄露到培养基中的情况下,包含无包膜病毒颗粒的培养上清可以用作样品。无包膜病毒生产细胞的破坏或裂解可以通过已知方法如超声处理、冻融处理、酶处理或渗透压处理实施。当由此获得的无包膜病毒生产细胞的溶解物或匀浆或包含无包膜病毒颗粒的培养上清具有中性或碱性ph时,溶解物或匀浆或培养上清可以直接用作包含无包膜病毒颗粒的中性或碱性样品。当溶解物或匀浆或培养上清不是中性或碱性ph,溶解物或匀浆或培养上清可以被中和以获得要在本发明方法中应用的包含无包膜病毒颗粒的中性或碱性样品。如本文所用的,中和意味着但不限于,将碱性溶液或ph缓冲液加入酸性溶液。中和后的溶液(中和产物)可为中性或碱性的。碱性溶液的例子包括但不限于氢氧化钠溶液和氢氧化钾溶液。ph缓冲液的例子包括但不限于tris-hcl溶液。
要在本发明方法中应用的包含无包膜病毒颗粒的中性或碱性样品也可以是通过用酸性溶液处理无包膜病毒生产细胞获得的提取物的中和产物。具体地,可以优选应用通过使无包膜病毒生产细胞接触酸性溶液而获得、并且然后被中和的样品。这一步骤通过将无包膜病毒生产细胞的团块(pellet)悬浮在酸性溶液中而实施,其中团块(pellet)通过细胞培养后离心或过滤而除去培养溶液而收集,或通过向无包膜病毒生产细胞的培养溶液加入成分而实施,其中该成分是能够使培养溶液为酸性的成分。酸性溶液没有限定,只要酸性溶液显示比培养中的无包膜病毒生产细胞的ph低的ph,并且酸性溶液使得能够从完成病毒生产的无包膜病毒生产细胞制备包含无包膜病毒颗粒的提取物。酸性溶液的例子包括与本发明方法步骤(b)中应用的酸性溶液的例子相同的溶液。能够使培养溶液为酸性的成分没有限定,只要该成分将无包膜病毒生产细胞的培养溶液的ph改变为比培养中的无包膜病毒生产细胞的ph低的ph,并且该成分使得能够从完成病毒生产的无包膜病毒生产细胞制备包含无包膜病毒颗粒的提取物。该成分的例子包括与本发明方法步骤(b)中应用的酸的例子相同的化合物。与酸性溶液或能够使培养溶液为酸性的成分接触的温度和时间的例子包括但不特别限于,0至40℃,优选地4至37℃,和1分钟至48小时,优选地5分钟至24小时。处于与酸性溶液或能够使培养溶液为酸性的成分接触的状态的无包膜病毒生产细胞、或处于中和后的状态的提取物也可以长期储存在超低温冰箱中,例如在-80℃。该接触步骤导致无包膜病毒释放到病毒生产细胞之外。
本发明方法中应用的包含无包膜病毒颗粒的中性或碱性样品可以预先用核酸酶处理。例如,在加入降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质之前,包含无包膜病毒颗粒的样品可以用核酸酶处理。用于该步骤的核酸酶优选地是作用于提取物中包含的dna的核酸酶。核酸酶的例子包括benzonase(注册商标)(由merckmilliporecorporation制造)和cryonase冷活化核酸酶(由takarabioinc.制造)。用于核酸酶处理的温度和时间没有特别限定,并且可以取决于要应用的核酸酶的种类适当确定。
在本发明方法中,除去形成的沉淀物以获得包含无包膜病毒颗粒的级分的步骤意味着,除去在酸化步骤中形成的沉淀物以获得包含无包膜病毒颗粒的级分的步骤。沉淀物可以通过已知的固液分离方法如过滤,优选地离心而除去。
通过除去沉淀物的步骤获得的包含无包膜病毒颗粒的级分可以进一步接受纯化步骤。通过纯化步骤,无包膜病毒颗粒可以被浓缩。用于纯化的方法的例子包括超速离心、层析、超滤和其它已知方法。
超滤意味着应用超滤膜过滤。超滤膜意味着具有物理上明确的许多细孔的分离膜。考虑到无包膜病毒的纯化和污染物质的除去,超滤膜的分子量边界(cutoff)优选地是50至300kda,更优选地70至150kda,进一步优选地100kda。例如,可以应用超滤膜amicon(注册商标)ultra-15,其具有100kda的分子量边界(由merckmilliporecorporation制造,下文称为100kda超滤膜)。同时,聚乙二醇(peg)可以加入含有无包膜病毒颗粒的级分以改善无包膜病毒颗粒的回收率。加入的peg没有特别限定,并且可以应用具有各种平均分子量的peg。例如,在本发明中应用具有200至10,000的平均分子量、优选地4,000至8,000的平均分子量的peg。更优选地,可以应用具有6,000的平均分子量的peg。
无包膜病毒颗粒通过层析的纯化可以通过离子交换柱(例如,mustangq,由pallcorp.制造)、亲和柱(例如,avbsepharose(注册商标),由gehealthcareltd.制造,或肝素柱)、羟基磷灰石柱等实施。
在本发明方法中,无包膜病毒颗粒优选地是腺伴随病毒颗粒。
在本发明方法中,无包膜病毒颗粒的收率显示为无包膜病毒的滴度等。无包膜病毒的滴度显示为但不限于,在一定量的样品中,(a)无包膜病毒的基因组的数目(基因组滴度),(b)如试验测定的无包膜病毒对细胞的感染能力(感染滴度),或(c)所测定的构成无包膜病毒的蛋白的纯度。
用于测定上述(a)的方法的例子包括包含通过pcr测定样品中病毒基因组的拷贝数的方法。对于基因组滴度的测定,应用例如aavpro(注册商标)滴定试剂盒(用于实时pcr)ver.2(由takarabioinc.制造),并且基因组滴度可以通过所附使用说明书中描述的方法计算。
用于测定上述(b)的方法的例子包括包含用系列稀释的无包膜病毒溶液感染合适的靶细胞和检测细胞的形状的变化(细胞变性)的方法、包含检测转基因的表达的方法、和包含测定引入细胞中的病毒基因组的拷贝数的方法。
用于测定上述(c)的方法的例子包括包含蛋白的sds-page分析的方法和包含通过免疫学技术定量测定蛋白的方法。
(2)用于本发明生产方法的试剂盒
本发明的试剂盒是用于实施上述部分(1)中描述的生产无包膜病毒颗粒的方法的试剂盒,并且包含:(1)降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质、和(2)酸性溶液。作为在酸性条件下沉淀的物质,可以优选地应用降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质,如上述部分(1)"本发明无包膜病毒颗粒的生产方法"中解释的。酸性溶液没有特别限定,只要其能够用于酸化样品的ph的步骤,所述样品中加入了降低在酸性条件下的蛋白的溶解度的物质和/或在酸性条件下沉淀的物质,如上述部分(1)"本发明无包膜病毒颗粒的生产方法"中解释的。本发明的试剂盒可以进一步包括中和溶液。中和溶液优选地是碱性溶液或ph缓冲液。碱性溶液的例子包括但不限于氢氧化钠溶液和氢氧化钾溶液。ph缓冲液的例子包括但不限于tris-hcl溶液。试剂盒可以进一步包含用于制备来自无包膜病毒生产细胞的提取物的试剂、包含提供对无包膜病毒的颗粒形成所必需的元件的核酸的质粒、包含要包封在无包膜病毒的颗粒中的核酸的质粒等。
根据本发明,除了生产无包膜病毒颗粒的方法,也提供用于该生产方法的试剂盒和通过该生产方法生产的无包膜病毒颗粒。通过应用本发明生产方法获得的无包膜病毒颗粒可以用作用于药物组合物的活性成分。该药物组合物可以离体(exvivo)应用于来自患者的细胞或直接施用给患者。
实施例
下文,通过实施例的方式对本发明进行更具体地解释,本发明不限于这些实施例。
实施例1:通过加入脱氧胆酸钠和加入柠檬酸纯化raav5
(1)用于raav的生产的细胞的接种
在包含10%fbs(由gibco制造)的dmem(由sigma-aldrichco.llc.制造)中,悬浮293t/17细胞。将悬浮液接种在组织培养处理的t225烧瓶(由corninginc.制造)中,然后,在5%(v/v)co2的co2培养箱中在37℃培养2天。
(2)用于raav5的生产的质粒的转染
通过应用磷酸钙方法,用各25µg的含有编码aav2rep蛋白和aav5cap蛋白的序列的prc5载体(由takarabioinc.制造)、含有编码腺病毒衍生e2a、va和v4的序列的phelper载体(由takarabioinc.制造)、和含有用于在aav2基因组的两个itrs之间的荧光蛋白zsgreen1的表达盒的paav-zsgreen1载体(由takarabioinc.制造)转染由实施例1-(1)获得的细胞。转染后6小时,将培养基完全除去,并且将40ml/烧瓶的含有2%胎牛血清(fbs)的dmem加入烧瓶。将细胞在5%(v/v)co2的co2培养箱中在37℃培养2天。
(3)通过柠檬酸缓冲液处理的提取物的制备
向由实施例1-(2)获得的烧瓶中,加入0.5ml的0.5medta(由wakopurechemicalindustries,ltd.制造)。将烧瓶在室温静置几分钟而剥离细胞。然后,收集含有细胞的培养溶液,在1,750×g在4℃离心10分钟。然后,除去上清。将细胞团块(pellet)悬浮在2ml柠檬酸缓冲液(38.1mm柠檬酸、74.8mm柠檬酸钠、75mm氯化钠、100mm氯化镁、ph5)中,通过涡旋混合器混合15秒,在室温静置5分钟,再次通过涡旋混合器混合15秒,然后在14,000×g在4℃离心10分钟以收集上清。向上清中加入1/10量的2mtris-hcl(ph9.5),并将由此获得的溶液用作提取物。
(4)通过加入脱氧胆酸钠和加入柠檬酸的沉淀物的形成和除去
向由实施例1-(3)获得的提取物中,加入1/100量的20u/μlcryonase(注册商标)冷活化核酸酶(由takarabioinc.制造)(最终浓度:0.2u/μl),然后在37℃反应1小时。将反应样品分为5个等分试样。向样品1中仅加入1/10量的1m柠檬酸。向样品2中仅加入1/10量的5%(v/v)脱氧胆酸钠。向样品3中加入1/10量的5%(v/v)脱氧胆酸钠,并且在样品于37℃静置30分钟后,进一步加入1/40量的1m柠檬酸。向样品4中加入1/10量的5%(v/v)脱氧胆酸钠,并且在样品于37℃静置30分钟后,进一步加入1/20量的1m柠檬酸。向样品5中加入1/10量的5%(v/v)脱氧胆酸钠,并且在样品于37℃静置30分钟后,进一步加入1/10量的1m柠檬酸。使每一样品接受ph测量。结果示于表1中。
在所有样品中,形成沉淀物。样品在5,000×g于4℃离心5分钟,并且收集上清以除去沉淀物。收集的上清用具有0.45μm孔径的由pvdf制成的压力驱动过滤装置millex(注册商标)(由merckmilliporecorp.制造,下文称为0.45μm孔径滤器)过滤以获得滤液作为浓缩前raav5(样品1-5)。
(5)浓缩前raav5的纯度的确认
将由实施例1-(3)获得的提取物和由实施例1-(4)获得的5种浓缩前raav5中的每一个与等量的5×样品缓冲液(由takarabioinc.制造)混合并于95℃保持10分钟。将每一混合物(4μl)应用在4-12%聚丙烯酰胺凝胶上并电泳。在完成电泳后,将凝胶浸入合适量的oriole荧光凝胶染色溶液(由bio-radlaboratories,inc.制造),并遮光振荡90分钟。振荡后,将凝胶用luminoshot400(由takarabioinc.制造)摄影。结果示于图1。在图1中,泳道c显示提取物,并且泳道1-5显示浓缩前raav5(样品1-5)。
图1中见到的所有条带是相应于污染蛋白的条带。由于浓缩前的阶段raav5的浓度低,没有检测到相应于raav5的条带。如图1中所见,与泳道c相比,泳道1和泳道2中所见的相应于污染蛋白的条带的数目低。这些结果显示,通过向raav5和污染蛋白的混合物加入柠檬酸或加入脱氧胆酸钠,污染蛋白可以沉淀和除去。如图1中所见,与泳道1和2相比,泳道3-5中所见的相应于污染蛋白的条带的数目低。这些结果显示,通过加入脱氧胆酸钠和加入柠檬酸的组合,污染蛋白可以更有效地沉淀和除去。此外发现,污染蛋白的除去效率取决于脱氧胆酸钠和柠檬酸的加入量而变化。具体地,泳道4和泳道5中所见的相应于污染蛋白的条带的数目非常低。因此,当以0.5%(v/v)的最终浓度加入脱氧胆酸钠并且加入柠檬酸使得成为ph4至6时,污染蛋白被有效除去。
(6)浓缩前raav5的回收率的测定
测定由实施例1-(3)获得的提取物和由实施例1-(4)获得的5种浓缩前raav5的基因组滴度。对于基因组滴度的测定,应用aavpro(注册商标)滴定试剂盒(用于实时pcr)ver.2(由takarabioinc.制造),并且按照试剂盒所附的说明书进行包括反应溶液的制备等的操作。基于由此获得的基因组滴度的数据,通过下式计算总基因组滴度。
式:总基因组滴度(vg)=基因组滴度(vg/ml)×总流量(ml)
假设提取物的总基因组滴度是100,相对于提取物的总基因组滴度的值(回收率)示于图2。在图2中,纵轴显示回收率(%),并且在横轴上,泳道c显示提取物,并且泳道1-5显示浓缩前raav5(样品1-5)。
如图2中所见,样品1-5的回收率是99%至113%,其接近100%。这些结果显示,当通过加入脱氧胆酸钠和加入柠檬酸纯化raav5时,raav5的回收率非常高。
图1和图2的结果显示,通过加入脱氧胆酸钠和加入柠檬酸的raav5的纯化是非常有效的纯化方法。具体地,通过加入脱氧胆酸钠和加入柠檬酸,污染蛋白可以沉淀和除去。尤其发现,当以0.5%(v/v)的最终浓度加入脱氧胆酸钠并且加入柠檬酸使得成为ph4至6时,raav5以高回收率纯化。
实施例2:raav5的纯化和浓缩
(1)脱氧胆酸钠和柠檬酸的加入量的研究
以与实施例1-(1)至(3)相同的方式,制备提取物。向提取物中加入1/100量的20u/μlcryonase(注册商标)冷活化核酸酶(由takarabioinc.)(最终浓度:0.2u/μl),然后在37℃反应1小时。将反应样品分为4个等分试样。向样品中加入1/10量或1/20量的5%(v/v)脱氧胆酸钠,并且在样品于37℃静置30分钟后,进一步加入1/20量或1/30量的1m柠檬酸。每一样品接受ph测量。结果示于表2。
在所有样品中,形成沉淀物。样品在5,000×g在4℃离心5分钟,并收集上清以除去沉淀物。用0.45μm孔径滤器过滤收集的上清以获得滤液作为浓缩前raav5。
(2)通过超滤膜的raav5的纯化和浓缩
将由实施例2-(2)获得的浓缩前raav5放在100kdak超滤膜(由merckmilliporecorp.制造)上,并在2,000×g在4℃离心10分钟。弃去滤液。进一步,将"一系列操作(洗涤操作),其中将10ml的d-pbs加在超滤膜上,悬浮和在上述条件下离心,并弃去滤液"重复5次以获得最终的浓缩物作为纯化raav5(样品a-d)。通过这一系列的操作,具有低分子量的污染物质被除去并且raav5被浓缩10倍。
(3)raav5的纯度的确认
以与实施例1-(5)相同的方式,确认由实施例2-(2)获得的纯化raav5的纯度。结果示于图3。在图3中,泳道m显示蛋白分子量标记(broad)(由takarabioinc.制造;从上面起200、116、97、66、44、29、20kda),并且泳道a-d显示纯化raav5(样品a-d)。
在图3中,3个粗条带是相应于raav5的结构蛋白(vp1、vp2、vp3)的条带,并且其它条带是相应于污染蛋白的条带。如从图3所见,泳道a具有低数目的相应于污染蛋白的条带。这一结果显示,通过除去加入脱氧胆酸钠(最终浓度0.5%(v/v))和加入柠檬酸(ph5)而形成的沉淀物,然后实施用100kdak超滤膜的处理,可以纯化和浓缩raav5。
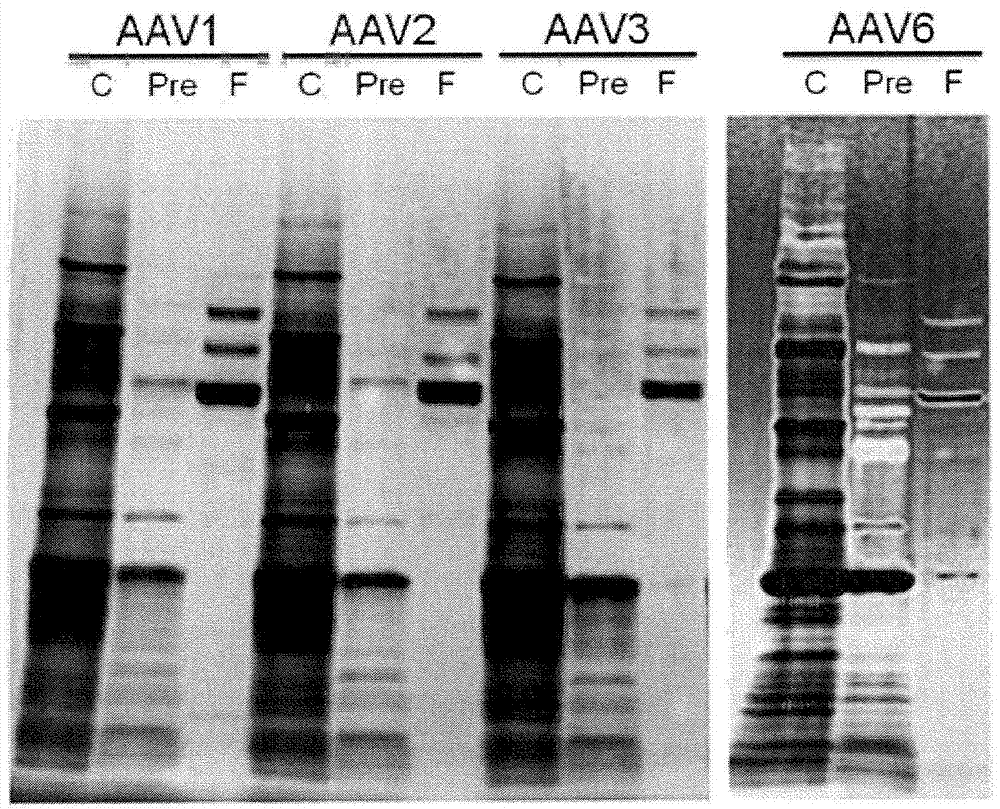
实施例3:各种血清型(raav1、raav2、raav3、raav6)的raav的纯化
(1)用于raav的生产的细胞的接种
以与实施例1-(1)相同的方式,接种用于raav的生产的细胞。
(2)用于raav的生产的质粒的转染
以与实施例1-(2)相同的方式,用用于raav的生产的质粒转染由实施例3-(1)获得的细胞,除了根据要制备的raav的血清型,应用prep-cap1aav辅助质粒(由appliedviromics制造)、prc2-mi342载体(由takarabioinc.制造)、prep-cap3aav辅助质粒(由appliedviromics制造)或prc6载体(由takarabioinc.制造)替代prc5载体。
(3)通过柠檬酸缓冲液处理的提取物的制备
以与实施例1-(3)相同的方式,制备每一血清型的raav的提取物。
(4)通过加入脱氧胆酸钠和加入柠檬酸的沉淀物的形成和除去
向由实施例3-(3)获得的提取物,加入1/100量的20u/μlcryonase(注册商标)冷活化核酸酶(由takarabioinc.制造)(最终浓度:0.2u/μl),然后在37℃反应1小时。然后,向每一样品加入1/10量的5%(v/v)脱氧胆酸钠。在样品于37℃静置30分钟后,向每一样品进一步加入1/20量的1m柠檬酸。每一样品接受ph测量。作为结果,每一样品具有ph4至6。
在所有样品中,形成沉淀物。样品在5,000×g在4℃离心5分钟,并收集上清以除去沉淀物。用0.45μm孔径滤器过滤收集的上清以获得滤液作为浓缩前raav。
(5)通过超滤膜的raav的纯化和浓缩
将由实施例3-(4)获得的浓缩前raav放在100kdak超滤膜上,并在2,000×g在4℃离心5分钟或更长时间。弃去滤液。进一步,将"一系列操作(洗涤操作),其中将14ml的d-pbs加在超滤膜上,悬浮和在上述条件下离心,并弃去滤液"重复5次以获得最终的浓缩物作为纯化raav。通过这一系列的操作,具有低分子量的污染物质被除去并且raav被浓缩3至5倍。
(6)raav的纯度的确认
以与实施例1-(5)相同的方式,确认每一血清型的raav的提取物(泳道c)、浓缩前raav(泳道pre)和纯化raav(泳道f)的纯度。结果示于图4。
在图4中,泳道c和泳道pre中所见的所有条带是相应于污染蛋白的条带。由于此阶段的raav浓度低,没有检测到相应于raav的条带。与之相比,泳道f中所见的3个粗条带是相应于raav的结构蛋白(vp1、vp2、vp3)的条带。如从图4所见的,关于每一血清型的raav,与泳道c相比,泳道pre具有低数目的相应于污染蛋白的条带。这一结果显示,通过加入脱氧胆酸钠和加入柠檬酸,可以沉淀和除去污染蛋白,不管raav的血清型如何。此外,关于每一血清型的raav,泳道f在相应于raav的结构蛋白(vp1、vp2、vp3)的条带之外几乎没有相应于污染蛋白的条带。这一结果显示,通过用100kdak超滤膜处理,raav可以被浓缩并且具有低分子量的污染蛋白可以被除去,不管raav的血清型如何。
(7)raav的回收率的测定
以与实施例1-(6)相同的方式,测定每一血清型的raav的基因组滴度。计算每一步骤的raav的总基因组滴度。假设提取物的总基因组滴度是100,相对于提取物的总基因组滴度的值(回收率)示于表3。
如图3中所见,浓缩前raav(pre)的回收率是89%至120%,其接近100%。这些结果显示,当通过加入脱氧胆酸钠和加入柠檬酸纯化raav时,raav的回收率非常高,不管raav的血清型如何。
图4和表3的结果显示,通过加入脱氧胆酸钠和加入柠檬酸的raav的纯化是非常有效的纯化方法,不管raav的血清型如何。具体地,通过以0.5%(v/v)的最终浓度加入脱氧胆酸钠并且加入柠檬酸使得成为ph4至6,污染蛋白可以沉淀和除去。进一步发现,通过额外实施用100kdak超滤膜的处理,以更高纯度获得raav。
实施例4:通过加入聚乙二醇改善raav6的回收率
(1)浓缩前raav6的制备
以与实施例3-(1)至(4)相同的方式,制备浓缩前raav6。
(2)通过超滤膜的raav6的纯化和浓缩(加入聚乙二醇)
将浓缩前raav6分为3个等分试样。样品1中不加入聚乙二醇。向样品2加入聚乙二醇(peg)-6000(由sigma-aldrichco.llc.制造)溶液以成为0.4%(w/v)的最终浓度。向样品3加入peg-600以成为0.8%(w/v)的最终浓度。将每一样品放在100kdak超滤膜上,并在2,380×g在室温离心10分钟或更长时间。弃去滤液。进一步,将"一系列操作(洗涤操作),其中将10至14ml的包含每一最终浓度的聚乙二醇的pbs加在超滤膜上,悬浮和在上述条件下离心,并弃去滤液"重复3次。仅样品2和样品3用pbs额外洗涤。获得最终的浓缩物作为纯化raav6。通过这一系列的操作,具有低分子量的污染物质被除去并且raav6被浓缩6至12倍。
(3)raav6的回收率的测定
以与实施例1-(6)相同的方式,测定每一raav6的基因组滴度。计算每一步骤的每一raav6的总基因组滴度。假设提取物的总基因组滴度是100,相对于提取物的总基因组滴度的值(回收率)示于表4。
如表4所见,在应用超滤膜纯化和浓缩raav6时,通过加入聚乙二醇,raav6的回收率被改善。认为在超滤时通过加入聚乙二醇,其它血清型的raav的回收率也可以被改善。
实施例5:各种酸性溶液的研究
(1)raav6的提取物的制备
以与实施例3-(1)至(3)相同的方式,制备raav6的提取物。
(2)通过加入脱氧胆酸钠和加入各种酸性溶液的沉淀物的形成和除去
向由实施例5-(1)获得的raav6提取物,加入1/100量的20u/μlcryonase(注册商标)冷活化核酸酶(由takarabioinc.制造)(最终浓度:0.2u/μl),然后在37℃反应1小时。将一部分反应样品分为3个等分试样。向每一样品加入1/10量的5%(v/v)脱氧胆酸钠,并在样品于37℃静置30分钟后,加入1/20量的1m柠檬酸、1/20量的1m盐酸或1/20量的1m乙酸。每一样品通过ph测量证实为酸性。每一样品的条件示于表5。
在所有样品中,形成沉淀物。样品在5,000×g在4℃离心5分钟,并收集上清以除去沉淀物。用0.45μm孔径滤器过滤收集的上清以获得滤液作为浓缩前raav6。
(3)浓缩前raav6的纯度和回收率的确定
以与实施例1-(5)相同的方式,确定浓缩前raav6的纯度。作为结果,浓缩前raav6具有相等的纯度,不管酸性溶液的种类如何。以与实施例1-(6)相同的方式,确定浓缩前raav6的基因组滴度并计算回收率。作为结果,浓缩前raav6具有相等的回收率,不管酸性溶液的种类如何。这些结果显示,当脱氧胆酸钠和酸性溶液加入raav和污染蛋白的混合物时,通过加入任何种类的酸性溶液,污染蛋白可以被沉淀和除去。
实施例6:各种表面活性剂的研究
(1)raav2的提取物的制备
以与实施例3-(1)至(3)相同的方式,制备raav2的提取物。
(2)通过加入各种表面活性剂和加入柠檬酸的沉淀物的形成和除去
向由实施例6-(1)获得的raav2提取物,加入1/100量的20u/μlcryonase(注册商标)冷活化核酸酶(最终浓度:0.2u/μl),然后在37℃反应1小时。将一部分反应样品分为3个等分试样。向每一样品加入1/10量的5%(v/v)脱氧胆酸钠、5%(v/v)鹅脱氧胆酸钠或5%(v/v)胆酸钠,并在样品于37℃静置30分钟后,进一步加入1/20量的1m柠檬酸。每一样品通过ph测量证实为酸性。每一样品的条件示于表6。
在所有样品中,形成沉淀物。样品在5,000×g在4℃离心5分钟,并收集上清以除去沉淀物。用0.45μm孔径滤器过滤收集的上清以获得滤液作为浓缩前raav2。
(3)通过超滤膜的raav2的纯化和浓缩
将由实施例6-(2)获得的浓缩前raav2放在100kdak超滤膜上,并在2,000×g在4℃离心5分钟或更长时间。弃去滤液。进一步,将"一系列操作(洗涤操作),其中将14ml的d-pbs加在超滤膜上,悬浮和在上述条件下离心,并弃去滤液"重复5次,以获得最终的浓缩物作为纯化raav2。通过这一系列的操作,具有低分子量的污染物质被除去并且raav2被浓缩。
(4)raav2的纯度的确定
以与实施例1-(5)相同的方式,确定raav2提取物(泳道c)、浓缩前raav2(泳道pre)和纯化raav2(泳道f)的纯度。结果示于图5。
在图5中,泳道c和泳道pre中所见的所有条带是相应于污染蛋白的条带。由于此阶段的raav2浓度低,没有检测到相应于raav的条带。与之相比,泳道f中所见的3个粗条带是相应于raav2的结构蛋白(vp1、vp2、vp3)的条带。如从图5所见的,关于所有表面活性剂,与泳道c相比,泳道pre具有低数目的相应于污染蛋白的条带。这一结果显示,通过加入表面活性剂和加入柠檬酸,可以沉淀和除去污染蛋白,不管表面活性剂的种类如何。
工业实用性
根据本发明的生产无包膜病毒的方法,可以不经辛苦操作得到具有高纯度的无包膜病毒颗粒。通过本发明的生产方法生产的无包膜病毒颗粒和包含该无包膜病毒颗粒作为活性成分的组合物在用于基因治疗的基础调查研究和临床应用领域中的基因转移方法中是非常有用的。
- 还没有人留言评论。精彩留言会获得点赞!