通过对选择性剪接进行适体介导的调节来实现的基因表达调控的制作方法
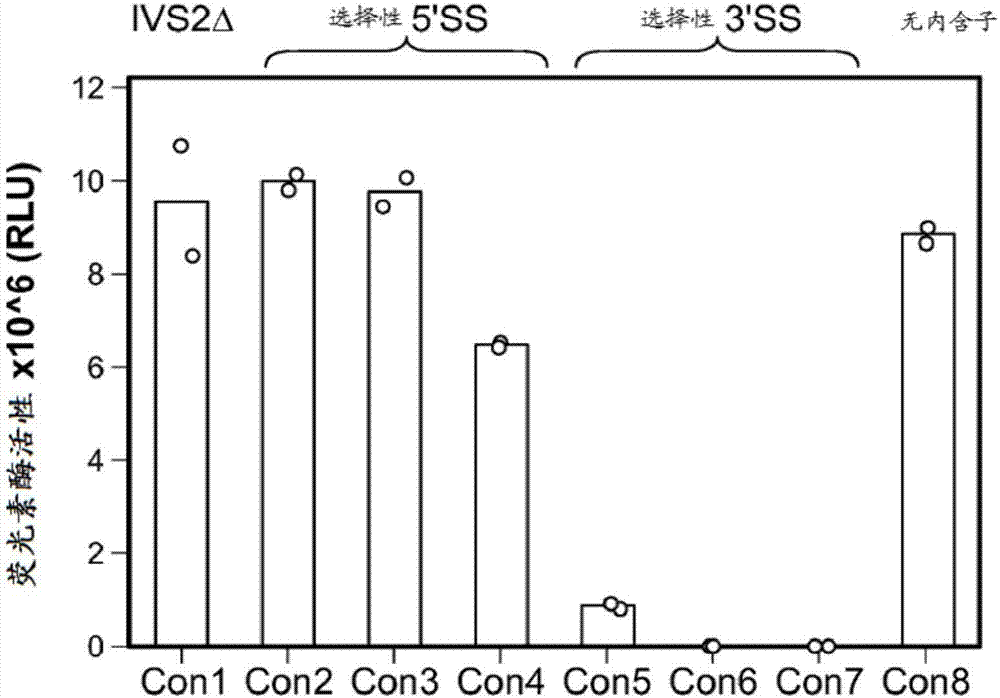
相关申请的交叉引用本申请要求2015年2月2日提交的美国申请号62/110,919的优先权,该申请以全文引用的方式并入本文中。发明领域本发明提供一种用于利用暴露于小分子来调控靶基因的表达的平台和使用所述平台的方法。所述平台以多核苷酸盒为特色,所述多核苷酸盒被放置于靶基因中,并且包括在5′内含子-选择性外显子-3′内含子背景下安置的合成核糖开关。核糖开关包含效应区和适体,所述适体结合配体(例如小分子)并且通过暴露于配体提供对靶基因表达的控制。发明背景剪接是指将内含子序列从新生前信使rna(前mrna)移除并且将外显子连接在一起形成mrna的过程。剪接位点为外显子与内含子之间的接合点,并且由内含子的5′和3′末端处的不同的一致序列来确定(即分别为剪接供体和剪接受体位点)。选择性前mrna剪接或选择性剪接为一种普遍的过程,所述过程存在于含有多个外显子的大多数人类基因中。它是通过称为剪接体的大多组分结构来进行,所述剪接体为小核核糖核蛋白(snrnp)和一系列不同的辅助蛋白的集合。通过识别各种顺式调控序列,剪接体确定外显子/内含子边界,移除内含子序列,并且将外显子剪接在一起成为最终的可翻译信息(即mrna)。在选择性剪接的情况下,可纳入或排除某些外显子以改变最终编码信息,从而改变所得到的被表达的蛋白。调控靶基因(例如治疗性转基因)的表达在多种情况下为必需的。在基因的治疗性表达的背景下,实现受调控的转基因表达的技术通过调控表达的水平和其定时而具有增强安全性的潜力。受调控而控制蛋白质表达的系统对于安全并且有效的治疗应用来说具有实际并且(在一些情况下)基本的作用。发明概要在一个方面中,本发明提供一种用于调控靶基因的表达的多核苷酸盒,所述多核苷酸盒包含(a)核糖开关和(b)选择性剪接的外显子,所述选择性剪接的外显子侧接5′内含子和3′内含子,其中核糖开关包含(i)效应区,所述效应区包含茎部,所述茎部包括3′内含子的5′剪接位点;以及(ii)适体,其中选择性剪接的外显子包含终止密码子,所述终止密码子在选择性剪接的外显子被剪接至靶基因mrna中时与靶基因同框。在一个实施方案中,适体特异性结合小分子配体。在一个实施方案中,用于调控靶基因的表达的多核苷酸(“基因调控盒”、“调控盒”或“多核苷酸盒”)含有5和3′内含子,所述内含子衍生自来自靶基因的内源性内含子。在一个实施方案中,5和3′内含子对靶基因来说是外源性的。在一个实施方案中,5和3′内含子衍生自人类β-球蛋白基因的内含子2。在一个实施方案中,5′内含子包含与靶基因同框的终止密码子。在一个实施方案中,5′和3′内含子的长度各自独立地为约50至约300个核苷酸。在一个实施方案中,5和3′内含子的长度各自独立地为约125至约240个核苷酸。在一个实施方案中,已对5′和/或3′内含子进行修饰以包括内含子剪接增强子、内含子剪接增强子、5′剪接位点、3′剪接位点或分支点序列,或改变其序列。在一个实施方案中,核糖开关的效应区茎部的长度为约7至约20个碱基对。在一个实施方案中,效应区茎部的长度为8至11个碱基对。在一个实施方案中,选择性剪接的外显子衍生自人类二氢叶酸还原酶基因(dhfr)的外显子2、突变体人类维尔姆斯瘤(wilmstumor)1外显子5、小鼠钙/钙调蛋白依赖性蛋白激酶iiδ外显子16或sirt1外显子6。在一个实施方案中,选择性剪接的外显子为来自seqidno:15的已修饰dhfr外显子2。在一个实施方案中,已以由以下组成的组中的一项或多项对选择性剪接的外显子进行了修饰:改变外显子剪接沉默子的序列、改变外显子剪接增强子的序列、添加外显子剪接增强子以及添加外显子剪接供体。在一个实施方案中,选择性剪接的外显子为合成的(即不衍生自天然存在的外显子)。在另一方面中,本发明提供一种调节靶基因的表达的方法,所述方法包括(a)将本发明的多核苷酸盒(如例如上文和本文中所描述)插入靶基因中,(b)将包含多核苷酸盒的靶基因引入细胞中,以及(c)将细胞暴露于小分子配体,所述小分子配体以有效诱导靶基因表达的量特异性结合适体。在一个实施方案中,当小分子配体存在时靶基因的表达比小分子配体不存在时的表达水平高约5倍以上。在一个实施方案中,当小分子配体存在时靶基因的表达比小分子配体不存在时的表达水平高约10倍以上。在一个实施方案中,将多核苷酸盒插入靶基因的蛋白质编码区中。在一个实施方案中,将两个或更多个多核苷酸盒插入靶基因中。在一个实施方案中,两个或更多个多核苷酸盒包含特异性结合于不同小分子配体的不同适体。在另一个实施方案中,两个或更多个多核苷酸盒包含特异性结合相同配体的不同适体的相同适体。在一个实施方案中,将包含多核苷酸盒的靶基因合并于用于靶基因的表达的载体中。在一个实施方案中,载体为病毒载体。在其他实施方案中,病毒载体选自由以下组成的组:腺病毒载体、腺相关病毒载体以及慢病毒载体。在另一方面中,本发明提供一种调节哺乳动物的眼睛中的靶基因的表达的方法,所述方法包括(a)向眼睛中引入包含靶基因的载体,所述靶基因含有多核苷酸盒,所述多核苷酸盒包含(i)核糖开关和(ii)选择性剪接的外显子,所述选择性剪接的外显子侧接5′内含子和3′内含子,其中合成核糖开关包含效应区,所述效应区包含茎部,所述茎部包括3′内含子的5剪接位点;以及适体,所述适体特异性结合配体,其中选择性剪接的外显子包含终止密码子,当选择性剪接的外显子被剪接至靶基因mrna中时所述终止密码子与靶基因同框;以及(b)以有效诱导靶基因的表达的量向哺乳动物提供配体。在一个实施方案中,配体为小分子。在一个实施方案中,通过眼内注射将载体引入眼睛。在一个实施方案中,载体为病毒载体。在一个实施方案中,病毒载体选自由以下组成的组:腺病毒载体、腺相关病毒载体以及慢病毒载体。在一个实施方案中,用于调控眼睛中靶基因的表达的多核苷酸含有5′和3′内含子,所述5′和3′内含子衍生自来自靶基因的内源性内含子。在一个实施方案中,5′和3′内含子对靶基因来说是外源性的。在一个实施方案中,5′和3′内含子衍生自人类β-球蛋白基因的内含子2。在一个实施方案中,5′内含子包含与靶基因同框的终止密码子。在一个实施方案中,5′和3′内含子的长度各自独立地为约50至约300个核苷酸。在一个实施方案中,5′和3′内含子的长度各自独立地为约125至约240个核苷酸。在一个实施方案中,已对5′和/或3′内含子进行修饰以包括内含子剪接增强子、内含子剪接增强子、5剪接位点、3′剪接位点或分支点序列,或改变其序列。在一个实施方案中,核糖开关的效应区茎部的长度为约7至约20个碱基对。在一个实施方案中,效应区茎部的长度为8至11个碱基对。在一个实施方案中,选择性剪接的外显子衍生自人类二氢叶酸还原酶基因(dhfr)的外显子2、突变体人类维尔姆斯瘤1外显子5、小鼠钙/钙调蛋白依赖性蛋白激酶iiδ外显子16或sirt1外显子6。在一个实施方案中,选择性剪接的外显子为来自seqidno:15的已修饰dhfr外显子2,即来自人类dhfr的已修饰外显子2。在一个实施方案中,已以由以下组成的组中的一项或多项对选择性剪接的外显子进行了修饰:改变外显子剪接沉默子的序列、改变外显子剪接增强子的序列、添加外显子剪接增强子以及添加外显子剪接供体。在一个实施方案中,选择性剪接的外显子为合成的(即不衍生自天然存在的外显子)。在一个方面中,本发明提供一种包含靶基因的重组多核苷酸,所述靶基因含有用于调控靶基因的表达(如例如上文所描述)的多核苷酸盒。在一个实施方案中,将多核苷酸盒定位于靶基因的蛋白质编码序列中。在一个方面中,本发明提供一种包含靶基因的载体,所述靶基因含有用于调控靶基因的表达(如例如上文所描述)的多核苷酸盒。在一个实施方案中,载体为病毒载体。在一个实施方案中,病毒载体选自由以下组成的组:腺病毒载体、腺相关病毒载体以及慢病毒载体。附图描述图1a.具有插入萤光素酶基因的编码序列中的截短的人类β-球蛋白内含子2(ivs2δ)的剪接构建体“con1”的示意性视图。名称“luci外显子1”和“luci外显子2”是指萤光素酶基因划分到5′和3′编码序列中的部分。对所插入的内含子序列ivs2δ的剪接产生全长mrna,所述全长mrna被翻译成全长蛋白质。图1b.内含子插入和剪接位点序列对萤光素酶表达的影响。con1至con7具有不同的内含子剪接位点(参见表1)。con1具有含有原始ivs25′剪接位点和3′剪接位点(分别为“5’ss”和“3’ss”)的ivs2δ。con2至con7具有插入的ivs2δ,但具有如表1中所列的5’ss和3’ss序列差异。con8不具有ivs2δ内含子。con1至con3展示与未插入内含子的萤光素酶对照(con8)相比内含子插入对萤光素酶表达无影响。具有较弱剪接位点的con4至con7展现降低的萤光素酶表达。图2a.具有所描绘的外显子纳入(i)和排除(ii)剪接模式的内含子-外显子-内含子盒的示意图。星形指示dhfr外显子2中的终止密码子。当选择性dhfr外显子因剪接而被纳入靶基因(萤光素酶)mrna中(i)时,所得转录物含有框内终止密码子,所述框内终止密码子阻断萤光素酶基因表达。仅在含有终止密码子的选择性dhfr外显子从最终mrna中排除时,靶基因才被表达(ii)。图2b.纳入或排除dhfr选择性外显子对萤光素酶基因的表达的影响。对用含有如图2a中所示的内含子-外显子-内含子盒的不同萤光素酶报告基因构建体转染的hek293细胞进行萤光素酶分析。将具有野生型剪接位点序列的dhfr外显子2(dhfr_wt)与在5′ss中含有突变(dhfr_wt5ssc)或用更强的con15’ss(dhfr_con15ss)或用来自con4的弱5’ss(dhfr_con45ss)代替天然5’ss的dhfr外显子相比较。在2b和2c中泳道1中所用的构建体为con1,在图1和实施例1中示出了con1。dhfr外显子2插入萤光素酶mrna使得萤光素酶表达降低,这在dhfr外显子2的5’ss(即剪接供体)突变时不发生(将dhfr_wt与dhfr_wt5ssc相比较)。当用来自con1的更强的5′ss代替dhfr外显子的5′ss时(构建体dhfr-con15′ss),dhfr外显子的纳入得以增强,从而产生与con1相比低545倍的萤光素酶表达。当使用弱5’ss(来自实施例1中的con4)来代替野生型5’ss时,此位点处的剪接减少,阻止了dhfr外显子纳入,从而使得萤光素酶表达增加。图2c.dhfr外显子序列内的外显子剪接增强子(ese)或抑制子(ess)元件影响选择性dhrf外显子的剪接并且调节靶基因的表达。dhfr外显子2内的srp40结合位点的突变使得萤光素酶表达显著降低:dhfr_wtmtsrp40表达与con1的表达之间相差2,982倍(图2cdhfr_wtmtsrp40;表2)。剪接增强子sc35的推定结合位点发生突变以产生更强的sc35结合位点(表2,strsc35)与con1相比进一步降低萤光素酶表达(139倍)(图2c,dhfr_wtstrsc35),这可能是由于dhfr外显子的纳入效率增加。用剪接抑制剂hnrnpa1的结合位点代替剪接增强子sc35的结合位点(表2,sc35hnrnpa1)使得萤光素酶表达与野生型dhfr外显子2相比增加4.3倍(图2c,dhfr_wtsc35hnrnpa1)。图3a.在选择性dhfr外显子2的5’ss处含有发夹结构的内含子-外显子-内含子盒的示意图。当dhfr5’ss嵌入发夹结构内时,dhfr外显子将不被纳入转录物中,因此允许萤光素酶被表达(x表示被埋在发夹中的dhfr外显子5’ss)。图3b.在图3a中所说明的内含子-外显子-内含子盒中所测试的四种不同发夹的序列和结构。图3c.dhfr外显子2的5’ss处的发夹结构对靶基因表达的影响。含有具有con15’ss序列的dhfr外显子的构建体因剪接的mrna中的dhfr外显子纳入而高效地抑制萤光素酶表达(dhfr_con15ss,图3c)。然而,dhfr外显子的5’ss嵌入发夹结构高效地阻止dhfr外显子的纳入,并且允许萤光素酶表达(dhfr_con15ss_hp15图3c)。具有被破坏的茎部的发夹序列不恢复萤光素酶表达(图3c.dhfr_con15ss_hp15x)。dhfr_wtmtsrp40构建体(实施例2)不表达萤光素酶,除非dhfr外显子的5’ss稳定地螯合于发夹结构中(dhfr_wtmtsrp40_hp15)。甚至在具有强剪接活性的突变体srp40结合位点的背景下(dhfr_wtmtsrp40_hp15x),发夹的去稳定化也阻止萤光素酶表达。图4a.和图4b.通过含有合成核糖开关的内含子-外显子-内含子调控盒进行基因调控的示意图。在不存在适体/配体结合的情况下,适体序列扰乱发夹茎部形成,使得dhfr外显子5’ss可接近,并且引起dhfr外显子的纳入,因此阻止翻译并且阻断蛋白质表达(图4a)。当发生适体/配体结合时,适体中的配体依赖性构象变化稳定茎部形成,从而螯合dhfr外显子5’ss,引起dhfr外显子排除和萤光素酶基因表达(图4b)。图4c.具有不同连接茎部长度的发夹茎部和茶碱适体构型。茶碱适体的茎部直接连接至发夹茎部,从而螯合dhfr外显子5’ss,产生20bp合成茎部。将茎部序列截短,从而产生具有不同茎部长度的一系列发夹。示出了dhfr_theo1、12、13以及14的茎部结构,茎部长度分别为20bp、9bp、8bp以及7bp。茶碱以符号(▲)表示。图4d.在存在和不存在茶碱的情况下使用茶碱适体的不同茎部长度对靶基因表达的影响。示出由如实施例4中所描述而产生的含有茶碱适体的调控盒(图4c)调控的萤光素酶表达的图。构建体theo1至12中,随着20bp的茎部长度降至9bp,发夹茎部具有足够的长度以在不存在适体/配体结合的情况下形成稳定结构。dhfr_theo13在不存在配体的情况下不形成稳定的发夹茎部,因此dhfr外显子5’ss未隐藏,从而引起dhfr外显子的纳入,阻断萤光素酶表达。在存在茶碱的情况下,发夹被稳定,并且dhfr外显子5’ss为剪接机械不可接近的。这引起dhfr外显子的排除,从而允许萤光素酶表达。在存在3mm茶碱的情况下,dhfr_theo_13相对于未诱导的基线表达水平展示43倍诱导。在存在或不存在茶碱的情况下,dhfr_theo_14不展示萤光素酶表达,表明甚至在适体结合至其配体时,此7bp茎部也太短而不能形成稳定发夹。因此,dhfr外显子被剪接至转录物中并且阻断萤光素酶表达。图5a.通过发夹茎部和适体p1茎部的连续截短产生的连接xpt-鸟嘌呤适体和dhfr外显子5’ss序列的合成茎部的序列。鸟嘌呤以符号(●)表示。图5b.茎部长度对核糖开关响应于适体配体结合而调控萤光素酶表达的能力的影响。将不同茎部长度的18个核糖开关插入调控盒中,并且将构建体转染至hek293细胞中,使所述hek293细胞在存在或不存在500μm鸟嘌呤的情况下生长。在不存在鸟嘌呤的情况下,构建体g14至g18与未调控的con1对照相比展示降低的萤光素酶表达。在鸟嘌呤存在下,萤光素酶表达在不同程度上得以恢复。图5c和5d.茎部长度对核糖开关响应于适体配体结合而调控萤光素酶表达的能力的影响的进一步分析。使用萤光素酶分析验证了构建体g11至g18。图5d示出了相对于con1的基础和诱导萤光素酶水平。在不存在鸟嘌呤的情况下,构建体g14至g17展示清楚的通过适体配体(本情况中为鸟嘌呤)对萤光素酶表达的调控。在不存在鸟嘌呤的情况下,萤光素酶表达水平低。在存在鸟嘌呤的情况下,萤光素酶表达被显著激活。图5d示出了这些受调控构建体在用鸟嘌呤诱导后所实现的占con1对照表达的百分比。图5e.在许多不同哺乳动物细胞类型中含有xpt-g17核糖开关的调控盒允许响应于配体而调控基因表达。将dhfr_g17转染至hepg2、aml12、rd以及c2c12细胞系中,并且在用鸟嘌呤处理后分析诱导的萤光素酶表达。显示与未将鸟嘌呤添加至细胞培养基时的未诱导基线表达水平相比当使细胞在配体存在下生长时对萤光素酶表达的倍数诱导。图5f.在病毒载体的背景下通过含xpt-g17调控盒对萤光素酶的调控。将具有含有xpt-g17调控盒的萤光素酶基因的构建体转移至aav载体骨架中并用于转染细胞。使细胞在存在和不存在鸟嘌呤的情况下生长。显示在存在鸟嘌呤的情况下对萤光素酶的倍数诱导,在用鸟嘌呤处理后,观察到1687倍表达诱导。图5g.通过调控盒响应于适体结合配体对抗体的调控。将具有xpt-g17核糖开关的内含子-外显子-内含子调控盒插入抗kdr抗体序列的前导肽序列中,并且将所得构建体转染至hek293细胞中。如通过elisa所分析,与未处理的细胞相比,在用配体处理转染细胞后存在80倍抗体蛋白质表达诱导。诱导的表达水平达到含有con1内含子序列的对照构建体的约40%。图5h.通过调控盒响应于适体/配体结合对所分泌的促红细胞生成素蛋白(epo)的调控。将具有xpt-g17核糖开关的内含子-外显子-内含子调控盒插入鼠类促红细胞生成素(epo)基因中,并且将所得构建体转染至hek293细胞中。如通过elisa所分析,在不存在鸟嘌呤的情况下观察到epo的低水平表达。在存在鸟嘌呤的情况下,观察到140倍epo表达诱导。图6a在调控盒中所测试的不同嘌呤核糖开关茎部的结构。嘌呤以符号●表示。图6b-6e.具有含有基于不同适体的核糖开关的调控盒的构建体的剂量反应(图6a中说明了核糖开关茎部)。图6b.含有鸟嘌呤适体的调控盒对鸟嘌呤的反应。图6c.含有鸟嘌呤适体的调控盒对鸟苷的反应。图6d.含有鸟嘌呤适体的调控盒对2’dg的反应。图6e.含有腺嘌呤适体的调控盒对腺嘌呤的反应。图7a.在含xpt-g17调控盒情况下通过鸟嘌呤对egfp的诱导。将具有xpt-g17核糖开关的内含子-外显子-内含子盒克隆至egfp基因中。将用构建体稳定转染的hek293细胞用500μm鸟嘌呤进行处理并且通过流式细胞术分析来分析处理后6h的gfp表达。鸟嘌呤处理使得egfp表达增加,图7a,(b)。图7b.靶基因表达对存在或不存在配体的反应。将用含有xpt-g17核糖开关的内含子-外显子-内含子盒稳定转染egfp的hek293细胞用500μm鸟嘌呤处理3天,并且每24h通过流式细胞术分析进行分析持续3天。将含鸟嘌呤培养基从细胞中洗去,并且使其在不存在鸟嘌呤处理的情况下继续再生长10天,并且监测egfp表达。当鸟嘌呤存在于细胞培养基中时egfp表达增加。在鸟嘌呤撤出时,egfp表达发生损失。图8a.通过两个拷贝的含xpt-g15调控盒调控的萤光素酶表达。该图示出了单个含xpt-g17或xpt-g15调控盒插入靶基因的构建体以及具有两个拷贝的含xpt-g15调控盒的构建体(xpt-g15双)的鸟嘌呤剂量反应。图8b.在组织培养细胞中通过不同调控盒调控的egfp表达。一个拷贝的含xpt-g17盒(egfp-xpt-g17)引起低的未诱导基线表达(a),并且与含有egfp-xpt-g15构建体的细胞相比达到较低的诱导表达水平(d)。一个拷贝的含xpt-g15盒(egfp-xpt-g15)得到较高的未诱导基线表达(b)以及较高的诱导表达(e)。在含有两个拷贝的含xpt-g15盒的构建体(egfp-xpt-g15双)的情况下,背景未诱导表达降低(c),而不降低诱导表达水平(f),因此倍数诱导增加。用鸟嘌呤处理细胞并且在处理后24h时成像。图8c.含有xpt-g15和xpt-g17核糖开关的调控盒对鸟嘌呤与鸟苷的反应。通过流式细胞术分析egfp表达的定量(平均荧光强度),并且以使用鸟嘌呤或鸟苷处理获得的平均荧光强度(mfi)除以未处理情况下所获得的mfi来计算倍数诱导。用鸟嘌呤和鸟苷处理给出类似的水平和倍数诱导。图8d.来自除一个拷贝的含ydhl-a5调控盒外含有一个拷贝的含xpt-g17调控盒的构建体的萤光素酶表达。将用此构建体转染的hek293细胞用鸟嘌呤或腺嘌呤或两者进行处理。在组合使用两种配体的情况下在其最高浓度下观察到最高萤光素酶诱导。图9a和9b.内含子截短对通过含有xpt-g17核糖开关的内含子-外显子-内含子调控盒调控的萤光素酶表达的影响。图9a示出了倍数诱导,而9b示出了与con1相比的萤光素酶表达百分比。图9c.相对于内含子-外显子-内含子调控构建体dhfr_g17缺失的序列的图。通过空心柱来描绘缺失的序列,通过实心柱来描绘其余序列。图9d和9e.图9c中所描绘的不同内含子缺失对基因调控的影响。内含子内侧接选择性dhfr外显子的序列改变外显子剪接和相对基因调控。举例来说,dhfr-g17_2ir_3展示内含子缺失,所述内含子缺失使得靶基因表达的倍数显著增加。图9d示出了倍数诱导,而图9e示出了相对于con1对照的绝对蛋白质表达水平。图10a和10b.不同外显子可在内含子-外显子-内含子调控盒中充当选择性外显子以调控基因表达。图10a显示具有不同外显子的构建体具有各种未诱导基线和诱导(500μm鸟嘌呤)萤光素酶表达水平。图10b示出了这些构建体情况下的诱导倍数,camkiid-e16产生与具有srp40激活突变的dhfr外显子(mtdhfr)等效的倍数诱导。图11a-c.在小鼠中在体内对萤光素酶表达的调控。通过流体力学注射将含有两个拷贝的xpt-g15调控盒(xpt-g15双,实施例8,图8a)的构建体递送至小鼠的肝脏。在dna递送后2h和12h时为小鼠经口给予不同剂量的鸟苷,并且然后成像。经口给予配体引起小鼠肝脏中被调控的靶基因的表达的剂量相关激活(图11a和图11b)。在另一个实验中,腹膜内施用鸟苷(图11c)。图像示出了100mg/kg或300mg/kg剂量的鸟苷处理前后的荧光素酶表达。在图(图11d)中,以平均光子/秒/mm2±标准偏差表示萤光素酶活性。(n=5)。图12.在小鼠视网膜中由基于核糖开关的aav载体介导的egfp转基因表达。示出在视网膜下注射后8天由aav2/8-gtx7在视网膜中介导的egfp转基因表达的荧光眼底照像(曝光时间:30s)。图13a和13b.视网膜下注射aav2/8-gtx7的单个鼠类眼睛的代表性眼底图像,这些图像示出视网膜中的egfp转基因表达随时间推移的变化。a-e:在载体注射后2、8、9、10以及12天时在白光照明下以200ms的曝光时间获取的图像。圆圈示出了通过瞳孔可见的视网膜区域,所述区域被视为roi用于定量。f-j:在475±25nm光照下以30s的曝光时间获取的图像,示出在载体注射后2、8、9、10以及12天时的egfp荧光。k-o:在载体注射后2、8、9、10以及12天时在475±25nm光照下以30s的曝光时间获取的图像,突出显示roi(圆圈)内超过强度阈值50的像素。图13b示出了诱导前(a)和诱导后(b)的高分辨率图像。图13c.在鼠类视网膜中在视网膜下注射aav2/8-gtx7之后随时间推移量化的egfp转基因表达。在以下时间点获取荧光眼底照片:视网膜下注射aav2/8-gtx7后2、8、9、10以及12天。曝光时间:30s,用于分析的像素强度阈值:50。在视网膜下注射aav2/8-gtx7后8、9以及10天时在成像之后进行腹膜内诱导。此外,在视网膜下注射aav2/8-gtx7后11天时进行玻璃体内诱导。基于单因素方差分析加上唐那氏校正(dunnettscorrection)来显示统计显著性,并且注射后8天作为对照点。图13d.在鼠类视网膜中视网膜下注射aav2/8-gtx5后随时间推移量化的egfp转基因表达(阳性对照)。在以下时间点获取获取荧光眼底照片:视网膜下注射aav2/8-gtx5后2、8、9、10以及12天。曝光时间:10s,用于分析的像素强度阈值:190。在视网膜下注射aav2/8-gtx5后8、9以及10天时在成像之后进行鸟苷的腹膜内施用。此外,在视网膜下注射aav2/8-gtx5后11天时进行鸟苷的玻璃体内施用。应用单因素方差分析加上邦弗朗尼校正(bonferronicorrection),并且在用鸟苷处理后在egfp表达中未发现统计显著性差异。详细说明本发明提供一种基因调控盒,所述基因调控盒包含在5′内含子-选择性外显子-3′内含子背景下的核糖开关。基因调控盒是指重组dna构建体,所述重组dna构建体当合并至靶基因的dna中时提供通过适体/配体介导的对所得前mrna的选择性剪接来调控靶基因的表达的能力。在本发明背景下的核糖开关含有感应区(例如适体)和效应区,所述感应区和效应区一起负责感应小分子配体的存在并且改变对选择性外显子的剪接。在一个实施方案中,靶基因的表达在适体配体存在时增加,而在配体不存在时降低。核糖开关如本文中所用的术语“核糖开关”是指rna多核苷酸的调控区段。在本发明背景下的核糖开关含有感应区(例如适体)和效应区,所述感应区和效应区一起负责感应配体(例如小分子)的存在并且改变对选择性外显子的剪接。在一个实施方案中,核糖开关为利用来自两个或更多个来源的多核苷酸重组的。如本文中所用的术语“合成”在核糖开关的背景下是指不天然存在的核糖开关。在一个实施方案中,感应区和效应区是通过多核苷酸接头连接。在一个实施方案中,多核苷酸接头形成rna茎部(即双链的rna多核苷酸的区域)。效应区在一个实施方案中,效应区包含3′内含子的5′剪接位点(“5’ss”)序列(即直接在选择性外显子的3′的内含子剪接位点序列)。效应区包含3′内含子的5′ss序列和与3′内含子的5′ss序列互补的序列。当适体结合其配体时,效应区形成茎部并且因此防止剪接至选择性外显子的3′末端处的剪接供体位点(参见例如图4b)。在某些条件下(例如当适体不结合于其配体时),效应区处于一种背景下,所述背景提供通往选择性外显子的3′末端处的剪接供体位点的通道,从而使得选择性外显子纳入靶基因mrna中(参见例如图4a)。效应区的茎部部分应具有足够的长度(和gc含量)以在配体结合适体后基本上防止选择性外显子的选择性剪接,而当配体不以足够的量存在时也允许到达剪接位点。在本发明的实施方案中,效应区的茎部部分除3′内含子的5′ss序列和其互补序列之外还包含茎部序列。在本发明的实施方案中,此额外茎部序列包含来自适体茎部的序列。可使用已知技术对茎部部分的长度和序列加以修饰,以识别当不存在配体时允许可接受的背景靶基因表达,而当配体存在时允许可接受的靶基因表达水平的茎部(参见例如实施例4和5以及图4c和4d、5a、5b、5c、5d)。如果茎部例如太长,那么它可能在存在或不存在配体的情况下隐藏通往3′内含子的5′ss序列的通道。如果茎部太短,那么它可能不会形成能够螯合3′内含子的5′ss序列的稳定的茎部,在这种情况下,在存在或不存在配体的情况下选择性外显子将被剪接至靶基因信息中。在一个实施方案中,效应区茎部的总长度在约7个碱基对至约20个碱基对之间。在一些实施方案中,茎部的长度在约8个碱基对至约11个碱基对之间。在一些实施方案中,茎部的长度为8个碱基对至11个碱基对。除了茎部的长度之外,可改变茎部的gc碱基对含量以改变茎部的稳定性。适体/配体如本文中所用的术语“适体”是指特异性结合于配体的rna多核苷酸。术语“配体”是指由适体特异性结合的分子。在一个实施方案中,配体为低分子量(小于约1,000道尔顿(dalton))分子,包括例如脂质、单糖、第二信使、其他天然产物和代谢物、核酸以及大多数治疗性药物。在一个实施方案中,配体为具有2个或更多个核苷酸碱基的多核苷酸。适体具有结合区,所述结合区能够与预期靶分子(即配体)形成复合物。结合的特异性可根据如与适体对不相关分子的解离常数相比较的适体对其配体的比较解离常数(kd)来定义。因此,配体为相较于不相关材料以更高的亲和力结合于适体的分子。通常,适体关于其配体的kd将比适体与不相关分子的kd小至少约10倍。在其他实施方案中,kd将小至少约20倍、小至少约50倍、小至少约100倍以及小至少约200倍。适体的长度将通常在约15与约200个核苷酸之间。更常见地,适体的长度将在约30与约100个核苷酸之间。可被合并作为核糖开关的一部分的适体可为天然存在的适体或其修饰型式,或被从头设计或通过指数式富集配体系统进化(selex)合成筛选的适体。结合小分子配体的适体的实例包括但不限于茶碱、多巴胺、磺酰罗丹明b(sulforhodamineb)以及纤维二糖卡那霉素a(cellobiosekanamycina)、利维霉素(lividomycin)、妥布霉素(tobramycin)、新霉素b、紫霉素、氯霉素、链霉素、细胞因子、细胞表面分子以及代谢物。关于识别小分子的适体的评述,参见例如famulok,science9∶324-9(1999)和mckeague,m.和derosa,m.c.j.nuc.aci.2012。在另一个实施方案中,适体为互补多核苷酸。在一个实施方案中,适体被设计成结合特定小分子配体。设计适体的方法包括例如selex。例如美国专利号5,475,096、5,270,163以及abdullahozer等人nuc.aci.2014中公开了使用selex设计选择性结合小分子的适体的方法,这些参考文献以引用的方式并入本文中。美国专利号5,580,737和5,567,588中描述了对selex方法的改良,这些专利以引用的方式并入本文中。用于识别适体的选择技术通常涉及制备大的含有被随机化或诱变的区域的所需长度的dna或rna分子的汇集物。举例来说,用于适体选择的寡核苷酸汇集物可含有具有20-100个随机化核苷酸的区,该区侧接约15-25个核苷酸长并且可用于结合pcr引物的规定序列的区。使用标准pcr技术或允许扩增所选核酸序列的其他手段来扩增寡核苷酸汇集物。当需要rna适体时可在体外对dna汇集物进行转录,以产生rna转录物的汇集物。然后基于特异性结合于所需配体的能力对rna或dna寡核苷酸的汇集物进行选择。选择技术包括例如亲和色谱法,不过可使用将允许基于特异性结合于另一分子的能力来选择核酸的任何方案。用于识别结合小分子并且在细胞内发挥功能的适体的选择技术可能涉及基于细胞的筛选方法。在亲和色谱法的情况下,使寡核苷酸与被固定于柱中的基质上或固定于磁珠上的靶配体接触。优选对寡核苷酸加以选择用于在盐浓度、温度以及模拟正常生理条件的其他条件存在的情况下进行配体结合。汇集物中结合于配体的寡核苷酸保留在柱或珠粒上,而非结合序列被洗掉。然后通过pcr(通常在洗脱后)对结合配体的寡核苷酸进行扩增(如果利用rna转录物,那么在逆转录之后进行)。对所选序列重复选择过程,进行总计约三至十轮的迭代选择程序。然后使用标准程序对所得寡核苷酸进行扩增、克隆以及测序以识别能够结合靶配体的寡核苷酸序列。一旦已识别适体序列,即可通过从包含诱变适体序列的寡核苷酸汇集物开始进行额外轮的选择来对适体进行进一步优化。可在一轮或多轮体外选择(例如selex)之后使用体内适体筛选。举例来说,konig,j.等人(rna.2007,13(4):614-622,以引用的方式并入本文中)描述了将selex与酵母三杂交系统组合用于适体的体内选择。选择性外显子作为本发明的基因调控多核苷酸盒的一部分的选择性外显子可为能够被转录为前mrna并且选择性剪接至靶基因的mrna中的任何多核苷酸序列。作为本发明的基因调控盒的一部分的选择性外显子含有至少一个抑制翻译的序列,使得当选择性外显子纳入靶基因mrna中时,阻止或减少由所述mrna引起的靶基因表达。在一个优选实施方案中,选择性外显子含有当选择性外显子因剪接而被纳入靶基因mrna中时与靶基因同框的终止密码子(tga、taa、tag)。在实施方案中,选择性外显子包含(除终止密码子外或作为终止密码子的替代物)当通过剪接将选择性外显子合并至靶基因mrna中时减少或基本上阻止翻译的其他序列,包括例如微rna结合位点,所述微rna结合位点导致mrna的降解。在一个实施方案中,选择性外显子包含mirna结合序列,所述mirna结合序列引起mrna的降解。在一个实施方案中,选择性外显子编码如下的多肽序列,所述多肽序列降低含有此多肽序列的蛋白质的稳定性。在一个实施方案中,选择性外显子编码如下的多肽序列,所述多肽序列引导含有此多肽序列的蛋白质进行降解。可通过改变外显子剪接增强子(ese)序列和外显子剪接抑制子(ess)序列和/或通过将ese或ess序列引入选择性外显子中来优化选择性外显子的基础或背景剪接水平。可使用本领域中已知的方法,包括但不限于定点诱变来实现对选择性外显子序列的此类改变。或者,可从商业来源获得所需序列(例如包含全部或部分的选择性外显子)的寡核苷酸并且克隆至基因调控盒中。可通过本领域中已知的方法,包括例如使用esefinder3.0(cartegni,l.等人esefinder:awebresourcetoidentifyexonicsplicingenhancers.nucleicacidresearch,2003,31(13):3568-3571)和/或其他可用资源来实现ess和ese序列的识别。在一个实施方案中,选择性外显子对靶基因来说为外源性的,不过它可衍生自来源于靶基因将在其中被表达的有机体的序列。在一个实施方案中,选择性外显子为合成序列(参见实施例10)。在一个实施方案中,选择性外显子为天然存在的外显子(参见实施例10)。在另一个实施方案中,选择性外显子衍生自全部或部分的已知外显子(参见实施例10)。在此背景下,“衍生”是指与天然存在的外显子基本上同源的含有选择性外显子的序列或其部分,但可含有各种突变,例如以引入将与靶基因序列同框的终止密码子,或引入或删除外显子剪接增强子和/或引入删除外显子剪接抑制子。在一个实施方案中,选择性外显子衍生自人类二氢叶酸还原酶基因(dhfr)的外显子2、突变体人类维尔姆斯瘤1外显子5、小鼠钙/钙调蛋白依赖性蛋白激酶iiδ外显子16或sirt1外显子6。5′和3′内含子序列选择性外显子侧接5和3′内含子序列。可用于本发明的基因调控盒中的5和3′内含子序列可为可从靶基因中剪接出来从而视存在或不存在结合适体的配体而定,形成靶基因mrna或在mrna中包含选择性外显子的靶基因的任何序列。5和3′内含子各自具有进行剪接所必需的序列,即剪接供体、剪接受体以及分支点序列。在一个实施方案中,基因调控盒的5和3′内含子序列衍生自一个或多个天然存在的内含子或其部分。在一个实施方案中,5和3′内含子序列衍生自截短人类β-球蛋白内含子2(ivs2δ)。在其他实施方案中,5′和3′内含子序列衍生自sv40mrna内含子(用于来自clonetech的pcmv-lacz载体中)、人类磷酸丙糖异构酶(tpi)基因的内含子6(nottajit等人rna.2003,9∶6070617)或来自人类因子ix的内含子(sumikokurachi等人j.bio.chem.1995,270(10),5276)、靶基因自己的内源性内含子或任何基因组片段或合成内含子(yilai等人humgenether.2006:17(10):1036),其含有足够的元件来进行调控剪接(thomasa.cooper,methods2005(37):331)。在一个实施方案中,本发明的选择性外显子和核糖开关被工程化为位于靶基因的内源性内含子中。也就是说,内含子(或基本上类似的内含子序列)天然存在于靶基因的该位置。在此情况下,紧接在选择性外显子上游的内含子序列被称为5′内含子或5′内含子序列,而紧接在选择性外显子下游的内含子序列被称为3′内含子或3′内含子序列。在此情况下,对内源性内含子进行修饰以含有侧接选择性外显子的5′和3′末端的剪接受体序列和剪接供体序列。可对本发明的基因调控盒中的剪接供体和剪接受体位点进行修饰以使其增强或削弱。也就是说,可通过标准克隆方法、定点诱变等对剪接位点进行修饰以更接近于对剪接供体或受体来说为一致的。更类似于剪接一致的剪接位点倾向于促进剪接并且因此为加强的。较不类似于剪接一致的剪接位点倾向于阻碍剪接并且因此为削弱的。对最常见类型的内含子(u2)的剪接供体来说一致的是a/cag||gta/gagt(其中||表示外显子/内含子界线)。对剪接受体来说一致的是cag||g(其中||表示外显子/内含子界线)。本领域中描述了在剪接供体和受体位点处特定核苷酸的频率(参见例如zhang,m.q.,hummolgenet.1988.7(5):919-932)。可调整5′ss和3′剪接位点的强度以调节选择性外显子的剪接。可对基因调控盒中的5′和3′内含子进行额外的修饰以调节剪接,包括修饰、删除和/或添加内含子剪接增强子元件和/或内含子剪接抑制子元件和/或改变分支位点序列。在一个实施方案中,已对5′内含子进行了修饰以含有终止密码子,所述终止密码子将与靶基因同框。还可对5′和3′内含子序列进行修饰以移除隐秘切割位点,所述隐秘切割位点可用公开可用的软件加以识别(参见例如kapustin,y.等人nucl.acidsres.2011.1-8)。可调整5′和3′内含子序列的长度以例如满足病毒表达构建体的尺寸要求。在一个实施方案中,5′和3′内含子序列的长度独立地为约50至约300个核苷酸。在一个实施方案中,5′和3′内含子序列的长度独立地为约125至约240个核苷酸。靶基因本发明的基因调控盒为可用于调控可在靶细胞、组织或有机体中表达的任何靶基因的表达的平台。术语“靶基因”是指被引入细胞中并且能够被转录至rna中并且在适当条件下翻译和/或表达的多核苷酸。或者,靶基因对靶细胞来说为内源性的,并且将本发明的基因调控盒安置到靶基因中(例如内源性靶基因的现有内含子中)。靶基因的一个实例为编码治疗性多肽的多核苷酸。在一个实施方案中,当使用本发明的基因调控盒来表达靶基因时,靶基因不被表达为包含选择性外显子的融合蛋白。选择性外显子的纳入因例如引起含有选择性外显子的信息的降解而使mrna的翻译最少化,或者例如归因于其过早截短而阻止功能性靶基因的表达。在一个实施方案中,靶基因对要在其中转录重组dna构建体的细胞来说为外源性的。在另一个实施方案中,靶基因为对要在其中转录重组dna构建体的细胞来说内源性的。在一个实施方案中,选择性外显子可含有与靶基因的编码序列同框的终止密码子。在其他实施方案中,选择性外显子可含有驱动转录物降解和/或阻断靶基因翻译的其他序列。根据本发明的靶基因可为编码蛋白质的基因,或编码非蛋白质编码rna的序列。靶基因可为例如编码结构蛋白、酶、细胞信号传导蛋白、线粒体蛋白、锌指蛋白、激素、转运蛋白、生长因子、细胞因子、细胞内蛋白、细胞外蛋白、跨膜蛋白、细胞质蛋白、核蛋白、受体分子、rna结合蛋白、dna结合蛋白、转录因子、翻译机构、通道蛋白、运动蛋白、细胞粘附分子、线粒体蛋白、代谢酶、激酶、磷酸酶、交换因子、伴侣蛋白以及这些中任一者的调节剂的基因。在实施方案中,靶基因编码促红细胞生成素(epo)、人类生长激素(hgh)、转录激活子样效应物核酸酶(talen)、人类胰岛素、crispr相关蛋白9(cas9)或免疫球蛋白(或其部分),包括例如治疗性抗体。表达构建体本发明涵盖使用重组载体来将编码靶基因并且含有本发明的基因调控盒的多核苷酸引入靶细胞中。在许多实施方案中,本发明的重组dna构建体包括额外dna元件,所述额外dna元件包括提供dna在宿主细胞中的复制以及靶基因在所述细胞中的适当水平的表达的dna区段。普通技术技工了解基于促进靶基因在靶细胞中的表达的能力来选择表达控制序列(启动子、增强子等)。“载体”意指包含要在体外或体内递送至宿主细胞中的多核苷酸的重组质粒、酵母人工染色体(yac)、微型染色体、dna微环或病毒(包括病毒衍生的序列)。在一个实施方案中,重组载体为病毒载体或多个病毒载体的组合。用于靶基因在靶细胞、组织或有机体中的表达的病毒载体为本领域中已知的,并且包括腺病毒(av)载体、腺相关病毒(aav)载体、逆转录病毒和慢病毒载体以及1型单纯疱疹(hsv1)载体。腺病毒载体包括例如基于人类2型腺病毒和人类5型腺病毒的那些,其已通过e1和e3区中的缺失而变为复制缺陷型。可将转录盒插入e1区中,从而产生重组e1/e3缺失av载体。腺病毒载体还包括辅助物依赖性高容量腺病毒载体(也称为高容量“无内脏”或“去内脏”载体),所述载体不含病毒编码序列。这些载体含有病毒dna复制和包装所需的顺式作用元件,主要为反向末端重复序列(itr)和包装信号(ψ)。这些辅助物依赖性av载体基因组具有携带从几百个碱基对至约36kb的外来dna的潜力。重组腺相关病毒“raav”载体包括衍生自任何腺相关病毒血清型的任何载体,包括但不限于aav-1、aav-2、aav-3、aav-4、aav-5、aav-7和aav-8、aav-9、aav-10等。raav载体可具有全部或部分缺失缺失的aav野生型基因中的一个或多个,优选为rep和/或cap基因,但保留功能性侧接itr序列。功能性itr序列被保留用于aav基因组的拯救、复制、包装以及潜在染色体整合。itr无需为野生型核苷酸序列,并且可改变(例如通过核苷酸的插入、缺失或取代),只要序列提供功能性拯救、复制以及包装即可。或者,可将诸如慢病毒载体等其他系统用于本发明的实施方案中。基于慢病毒的系统可转导非分裂以及分裂细胞,使它们可用于靶向例如cns的非分裂细胞的应用。慢病毒载体衍生自人类免疫缺陷病毒,并且像所述病毒一样,整合至宿主基因组中,从而提供非常长期的基因表达的潜力。还可使用例如阳离子脂质、聚合物或两者作为载体通过非病毒载体系统将携带含有基因调控盒的靶基因的包括质粒、yac、微型染色体以及微环的多核苷酸引入细胞或有机体中。还可使用共轭聚l-赖氨酸(pll)聚合物和聚乙烯亚胺(pei)聚合物系统来将载体递送至细胞。针对细胞培养物与有机体,用于将载体递送至细胞的其他方法包括流体力学注射和电穿孔以及使用超声波。关于用于基因递送的病毒和非病毒递送系统的评述参见以引用的方式并入本文中的nayerossadat,n.等人(advbiomedres.2012;1∶27)。调节靶基因的表达的方法在一个方面中,本发明提供一种通过(a)将本发明的基因调控盒插入靶基因中;(b)将包含基因调控盒的靶基因引入细胞中;并且(c)将细胞暴露于结合适体的配体来调节靶基因(例如治疗性基因)的表达的方法。在一个实施方案中,配体为小分子。在多个方面中,靶基因在靶细胞中的表达赋予将其引入其中的细胞所需性质,或以其他方式产生所需治疗性结果。在一个优选实施方案中,将基因调控盒插入靶基因的蛋白质编码序列(而不是5′或3′未翻译区域)中。在一个实施方案中,将单个基因调控盒插入靶基因中。在其他实施方案中,将2、3、4或更多个基因调控盒插入靶基因中。在一个实施方案中,将两个基因调控盒插入靶基因中。当将多个基因调控盒插入靶基因中时,它们各自可含有相同适体,使得可在多个盒处使用单个配体来调节选择性剪接并且从而调节靶基因表达。在其他实施方案中,将多个基因调控盒插入靶基因中,所述多个基因调控盒各自可含有不同适体,使得暴露于多个不同小分子配体来调节靶基因表达。在其他实施方案中,将多个基因调控盒插入靶基因中,所述多个基因调控盒各自含有不同的5’内含子、选择性外显子以及3’内含子序列。这在减少重组和改善合并至病毒载体中的容易性方面可为有用的。处理方法和药物组合物本发明的一个方面提供一种调控通过基因治疗递送的治疗性蛋白质的水平的方法。在此实施方案中,“靶基因”可编码治疗性蛋白质。“靶基因”可编码对细胞来说内源或外源的蛋白质。例如通过载体将含有具有适体驱动的核糖开关的调控盒的治疗性基因序列递送至体内的靶细胞。“靶基因”的细胞特异性可受启动子或载体内的其他元件控制。含有靶基因的载体构建体的递送以及对靶组织进行转染从而产生被调控靶基因的稳定转染是产生治疗性蛋白质的第一步。然而,由于靶基因序列内存在调控盒,靶基因不以显著水平表达,即在不存在结合于调控盒核糖开关内所含的适体的特异性配体的情况下它处于“关闭状态”。仅当施用适体特异性配体时,靶基因表达才被激活。含有靶基因的载体构建体的递送和激活配体的递送通常在时间上是隔开的。激活配体的递送将控制靶基因何时被表达以及蛋白质表达的水平。可通过许多途径来递送配体,包括但不限于经口、肌肉内(im)、静脉内(iv)、眼内或局部。配体的递送时间将取决于激活靶基因的要求。举例来说,如果持续需要由靶基因编码的治疗性蛋白质,那么可每天或一天多次递送经口小分子配体,以确保靶基因的持续激活,并且因此确保治疗性蛋白质的持续表达。如果靶基因具有长效作用,那么可较不频繁地给予诱导配体。本发明允许以由对调控盒内的适体具特异性的配体的按时给予决定的方式在时间上控制治疗性转基因的表达。治疗性转基因仅在配体施用后表达通过允许靶基因在不存在配体的情况下关闭而增加基因治疗处理的安全性。可使用不同的适体使不同的配体激活靶基因。在本发明的某些实施方案中,含有调控盒的各治疗性基因将在盒内具有特异性适体,所述特异性适体将由特异性小分子激活。这意味着各治疗性基因仅能被对其中所容纳的适体具特异性的配体激活。在这些实施方案中,各配体将仅激活一种治疗性基因。这使得有可能可将若干不同的“靶基因”递送至一个个体,并且各自将在递送对各靶基因中所容纳的调控盒内所含的适体具特异性的配体后被激活。本发明允许在递送激活配体时由身体产生基因可被递送至体内的任何治疗性蛋白质(诸如促红细胞生成素(epo)或治疗性抗体)。此治疗性蛋白质递送方法可代替在身体外部制造此类治疗性蛋白质然后注射或输注所述治疗性蛋白质,例如用于癌症中或用于阻断炎性或自身免疫疾病的抗体。含有受调控的靶基因的身体变成生物制剂制造工厂,当施用基因特异性配体时,所述生物制剂制造工厂开启。治疗性蛋白质的给药水平和给药时间对治疗效果来说可为关键的。举例来说,在用于癌症的(抗vegf抗体)的递送中。本发明增加了响应于对治疗性蛋白质水平和效果的监测进行给药的容易性。在一个实施方案中,靶蛋白质可为可靶向和编辑特定dna序列的核酸酶。此类核酸酶包括cas9、含锌指核酸酶或talen。在这些核酸酶的情况下,核酸酶蛋白质可能仅需要足以编辑靶内源性基因的短的时间段。然而,如果将未调控的核酸酶基因递送至体内,那么在细胞的剩余寿命中此蛋白质可能都存在。在核酸酶的情况下,核酸酶存在的时间越长,存在脱靶编辑的风险越高。此类蛋白质的表达的调控具有显著的安全性优点。在此情况下,可将含有含调控盒的核酸酶靶基因的载体递送至体内适当的细胞。在不存在盒特异性配体的情况下,靶基因处于“关闭”状态,因此不产生核酸酶。仅在施用激活配体时,才产生核酸酶。当经过足够的时间,允许发生足够的编辑时,将配体撤出并且不再施用。因此,核酸酶基因因而处于“关闭”状态,并且不再产生核酸酶,并且编辑停止。可使用此方法来克服遗传病状,包括许多遗传性视网膜病变,诸如由cep290的突变引起的lca10以及由abca4的突变引起的斯特格氏病(stargardtsdisease)。可利用仅在特异性配体施用后被激活的编码治疗性蛋白质的受调控靶基因的施用来调控治疗性基因以治疗许多不同类型的疾病,例如用治疗性抗体治疗癌症,用免疫调节蛋白或抗体治疗免疫病症、代谢疾病、用抗c5抗体或抗体片段作为受调控基因治疗诸如pnh等罕见疾病或用治疗性抗体治疗眼血管生成,以及用免疫调节蛋白治疗干性amd。允许治疗多种特定疾病和病状的多种特异性靶基因适合用于本发明中。举例来说,可使用胰岛素或胰岛素类似物(优选人类胰岛素或人类胰岛素类似物)作为治疗i型糖尿病、ii型糖尿病或代谢综合征的靶基因;可使用人类生长激素作为治疗具有生长障碍的儿童或缺乏生长激素的年人的靶基因;可使用促红细胞生成素(优选人类促红细胞生成素)作为治疗因慢性肾病引起的贫血、因脊髓发育不良引起的贫血或因癌症化疗引起的贫血的靶基因。本发明可特别适合用于治疗由单一基因缺陷引起的疾病,诸如囊性纤维化、血友病、肌肉萎缩症、地中海贫血或镰状细胞性贫血。因此,可使用人类β-球蛋白、γ-球蛋白、δ-球蛋白或ζ-球蛋白作为治疗β-地中海贫血或镰状细胞性贫血的靶基因;可使用人类因子viii或因子ix作为治疗血友病a或血友病b的靶基因。通常将本发明中所用的配体与一种或多种药学上可接受的载体组合以形成适合向患者施用的药物组合物。药学上可接受的载体包括通常用于制药领域中的溶剂、粘合剂、稀释剂、崩解剂、润滑剂、分散介质、涂料、抗细菌和抗真菌剂、等渗和吸收延迟剂等。药物组合物可呈片剂、丸剂、胶囊、锭剂等形式,并且被配制成与其预期施用途径相容。施用途径的实例包括肠胃外,例如静脉内、真皮内、鼻内、皮下、经口、吸入、经真皮(局部)、经粘膜以及经直肠。以一种给药方案向患者施用包含配体的药物组合物,使得足以适当调控靶基因的量的配体被递送至患者。当配体为小分子并且剂型为片剂、丸剂等时,优选地,药物组合物包含0.1mg至10g的配体;0.5mg至5g的配体;1mg至1g的配体;2mg至750mg的配体;5mg至500mg的配体;或10mg至250mg的配体。可每天一次或每天多次(例如每天2、3、4、5或更多次)给予药物组合物。或者,可少于每天一次给予药物组合物,例如每2、3、4、5、6、7、8、9、10、11、12、13或14天一次或每月一次或每几个月一次。在本发明的一些实施方案中,可仅少数的几次向患者施用药物组合物,例如一次、两次、三次等。本发明提供一种治疗需要增加由靶基因编码的治疗性蛋白质的表达的患者的方法,所述方法包括向患者施用包含针对适体的配体的药物组合物,其中先前已为患者施用了包含靶基因的重组dna,其中靶基因含有本发明的基因调控盒,所述本发明基因调控盒提供由适体的配体通过靶基因的前mrna的选择性剪接调控靶基因的表达,从而增加治疗性蛋白质的表达的能力。制品和试剂盒还提供用于本文中所描述的方法中的试剂盒和制品。在多个方面中,试剂盒包含在适合的包装中的本文中所描述的组合物(例如用于递送包含含有基因调控盒的靶基因的载体的组合物)。适合于本文中所描述的组合物(诸如注射用眼用组合物)的包装为本领域中已知的,并且包括例如小瓶(诸如密封小瓶)、容器、安瓿、瓶、罐、软包装(例如密封的麦拉(mylar)或塑料袋)等。可进一步对这些制品进行灭菌和/或密封。本发明还提供包含本文中所描述的组合物的试剂盒,并且可进一步包含关于使用组合物的方法,诸如本文中所描述的用途的说明。本文中所描述的试剂盒可进一步包括从商业和用户观点来看理想的其他材料,包括其他缓冲液、稀释剂、过滤器、针头、注射器以及带有用于进行施用(包括例如本文中所描述的任何方法)的说明的包装插页。举例来说,在一些实施方案中,试剂盒包括用于包含本发明的基因调控盒的靶基因的表达的raav、适合于注射的药学上可接受的载体以及以下中的一种或多种:缓冲液、稀释剂、过滤器、针头、注射器以及带有用于进行注射的说明的包装插页。在一些实施方案中,试剂盒适合用于眼内注射、肌肉内注射、静脉内注射等。如本文中所用的“同源性”和“同源的”是指在两个多核苷酸序列之间或在两个多肽序列之间的同一性百分比。一个序列与另一个序列之间的对应关系可通过本领域中已知的技术来确定。举例来说,可通过比对序列信息并且使用可轻易获得的计算机程序直接比较两个多肽分子来确定同源性。或者,可如下确定同源性:使多核苷酸在同源区域之间形成稳定双链体的条件下杂交,随后用单链特异性核酸酶消化,并且测定消化片段的尺寸。当在最佳比对而具有适当插入或缺失之后,如使用上述方法所确定,在确定长度的分子上分别至少约80%、至少约85%、至少约90%以及至少约95%的核苷酸或氨基酸匹配时,两个多核苷酸或两个多肽序列为彼此“基本上同源的”。相对于参考多肽或核酸序列的“序列同一性百分比”定义为在比对序列并且引入空隙(如果有必要)以实现最大序列同一性百分比之后,候选序列中与参考多肽或核酸序列中的氨基酸残基或核苷酸同一的氨基酸残基或核苷酸的百分比。出于确定氨基酸或核酸序列同一性百分比的目的而进行的比对可以普通技术技工已知的方式来实现,例如使用公开可用的计算机软件程序,包括blast、blast-2、align以及megalign(dnastar)软件。如本文中所用的术语“多核苷酸”或“核酸”是指任何长度的核苷酸(核糖核苷酸或脱氧核糖核苷酸)聚合形式。因此,此术语包括但不限于单链、双链或多链dna或rna、基因组dna、cdna、dna-rna杂交体或包含嘌呤和嘧啶碱基或其他天然、化学或生物化学修饰、非天然或衍生核苷酸碱基的聚合物。“异源的”或“外源的”意指衍生自基因型不同于相比较或引入或合并至其中的实体的其余部分的实体。举例来说,通过基因工程技术引入不同细胞类型的多核苷酸为异源多核苷酸(并且当被表达时可编码异源多肽)。类似地,合并至病毒载体中的细胞序列(例如基因或其部分)相对于载体为异源核苷酸序列。应了解并且预期,本领域技术人员可对本文中所公开的本发明原理作出改变,并且此类修改指在被包括在本发明范围内。以下实施例进一步说明本发明,但不应被解释为以任何方式限制本发明的范围。本文中所引用的所有参考文献以全文引用的方式并入本文中。实施例实施例1.剪接位点强度对受调控基因的表达的影响实验程序质粒构建体:luci-bsai-受体:通过限制酶spei和noti使含有cmv启动子的dna片段从phage-cmv-egfp-w(harvarduniversity)载体释放,并且将此片段克隆至用spei和noti消化的phdm-g(harvarduniversity)载体中。通过用bsmi和bstxi消化,移除3’突出物并且连接将所得载体中含有sv40ori序列的片段删除。对后续载体进行定点诱变(agilent)以删除ampr基因中的bsai位点。然后将所得载体用noti和bamhi消化,并且与含有noti-bsai-bamhi位点的片段连接以产生最终luci-bsai-受体载体。使用phdm-g作为含有被认为对剪接来说不关键的中间部分的缺失的人类β-球蛋白内含子2(“ivs2δ”)的模板(参见表5,seqidno:1)。使用pgl3-启动子(promega)作为萤火虫萤光素酶基因的模板。构建体8:使用引物luc-for-bsai和luci-rev-bsai通过pcr对萤光素酶基因进行扩增。将pcr产物用bsai消化并且克隆至bsai消化的luci-bsai-受体载体中。通过将3bsai消化的pcr产物连接至bsai消化的luci-bsai-受体中来制备剪接构建体1-7(con1-7,seqidno.1-seqidno.7),所述剪接构建体1-7表达插入有内含子ivs2δ的萤光素酶基因,所述内含子ivs2δ在内含子序列的各末端处具有不同的5′ss和3′ss。使用pgl3-启动子载体作为萤光素酶模板,并且使用phdm-g作为ivs2δ的模板。对于con1-7,用于扩增pcr片段的引物对如下:con1:luci-for-bsai/luci-剪接物-rev_1、ivs2-bsai-for/ivs2-bsai-rev_1以及luci-剪接物-for_1/luci-rev-bsai;con2:luci-for-bsai/luci-剪接物-rev_2、ivs2-bsai-for/ivs2-bsai-rev_2以及luci-剪接物-for_2/luci-rev-bsai;con3:luci-for-bsai/luci-剪接物-rev_3、ivs2-bsai-for/ivs2-bsai-rev_3以及luci-剪接物-for_3/luci-rev-bsai;con4:luci-for-bsai/luci-剪接物-rev_4、ivs2-bsai-for/ivs2-bsai-rev_1以及luci-剪接物-for_4/luci-rev-bsai;con5:luci-for-bsai/luci-剪接物-rev_1、ivs2-bsai-for/ivs2-bsai-rev_1以及luci-剪接物-for_5/luci-rev-bsai;con6:luci-for-bsai/luci-剪接物-rev_1、ivs2-bsai-for/ivs2-bsai-rev_1以及luci-剪接物-for61/luci-rev-bsai;con7:luci-for-bsai/luci-剪接物-rev_1、ivs2-bsai-for/ivs2-bsai-rev_1以及luci-剪接物-for71/luci-rev-bsai。通过dna测序验证所有构建体。表1.剪接构建体(con1-7)的剪接位点。由||标记内含子/外显子边界。构建体5′剪接位点3′剪接位点con1agg||gtgagttcttatcttcctcccacag||ccon2aaa||gtaagctcttatcttcctcccacag||ccon3gca||gtaagttcttatcttcctcccacag||ccon4gag||gtgtggtcttatcttcctcccacag||ccon5agg||gtgagtctttacttctatgactgtag||ccon6agg||gtgagtgtgactgtgtgtatgcacag||ccon7agg||gtgagtattgtgatcgcagccaatag||c转染:在转染前一天,将3.5x10^4个hek293细胞接种在96孔平底板中。将质粒dna(500ng)添加至管或96孔u形底板中。单独地,将transit-293试剂(mirus;1.4ul)添加至50μlopti-memi培养基(lifetechnologies)中,并且使其在室温(rt)下静置5分钟。然后,将50ul此稀释转染试剂添加至dna中,混合并且在室温(“rt”)下孵育20min。最后,将7μl此溶液添加至96孔板中的细胞孔中。所培养细胞的萤火虫萤光素酶分析:培养基更换后24小时,将板从孵育器中移出,并且在实验台上平衡至rt若干分钟,然后抽吸。添加glo-lysis缓冲液(promega,100μl,rt),并且使板在rt下保持至少5分钟。然后,通过50μl研磨混合孔内容物,并且将20μl的各样品与已在glo-lysis缓冲液中稀释至10%的20μlbright-glo试剂(promega)混合。使96个孔在不透明白色384孔板上间隔开。在rt下孵育5min之后,以500毫秒读取时间使用tecan机器测量发光。以平均相对光单位(rlu)±标准偏差表示萤光素酶活性。结果为了建立基于剪接的基因调控平台,我们首先测试了(i)将内含子插入感兴趣的基因(本情况中为萤火虫萤光素酶基因)的编码序列(cds)中对基因表达的影响(图1a)以及(ii)不同的5’ss和3’ss对基因表达的影响。将在各末端处具有不同的5’ss和3’ss的截短人类β-球蛋白内含子2(ivs2δ)插入萤火虫萤光素酶基因的编码序列中以测试剪接效率。构建体con8不具有ivs2δ内含子,而con1(seqidno.:1)具有带有原始ivs25’和3’ss序列的ivs2δ。构建体con2至con7(seqidno.:2-7)具有带有如表1中所列的不同的5’和3’ss序列的ivs2δ。如图1b中所示,具有天然ivs2剪接位点的ivs2δ插入萤光素酶基因中不影响基因表达(将con1与con8相比较)。然而,用具有不同强度的剪接位点序列代替ivs2δ中的ivs2剪接位点显著削弱萤光素酶表达。如图1b中所示,5’ss改变了的con2和con3具有类似于con1和con8的表达水平,然后con4中的5’ss改变以及con5至con7中的3’ss改变不显著降低萤光素酶表达(将con4至con7与con8相比较)。因此,剪接位点的差异影响靶基因表达。使用con1来进行进一步研发。实施例2.内含子-外显子-内含子盒和顺式元件对剪接在调节靶基因表达中的影响.实验程序使用esefinder3.0预测推定外显子剪接增强子(ese)序列。合成(idt)具有内含子侧接序列的野生型人类二氢叶酸还原酶(dhfr)外显子2,所述内含子侧接序列具有天然5’ss(dhfr-wt;(表2);seqidno.:47)、四个核苷酸突变至c的天然5’ss(dhfr-wt5ssc;(表2);seqidno.:48)、来自con1(dhfr-con15ss;表2seqidno.:49)或con4(dhfr-con45ss,seqidno.:50)的5’ss序列。为测试dhfr外显子2内的ese和外显子剪接抑制子(ess)序列的作用,合成了不同的dhfr外显子2突变体(列于表2中)。使用金门克隆策略(goldengatecloningstrategy,neb)将所有这些不同的dhfr外显子2序列克隆至con1构建体中的ivs2δ内含子的近似中心。通过dna测序(genewiz)验证构建体。在hek293细胞中转染dna并且如实施例1中所描述分析萤光素酶活性。表2.含有修饰过的剪接调控序列的dhfr外显子2。加下划线的序列表示dhfr外显子2内的修饰过的剪接调控序列。结果将野生型人类dhfr外显子2和相邻的内含子序列(seqidno.:8)插入con1构建体中的ivs2δ内含子的近似中心。此构型产生一种平台,其中靶基因的内含子序列中的外源性外显子可充当选择性外显子,从而允许通过调节选择性外显子剪接来调控靶基因的表达。在此构型(图2a)中,可在靶基因的5′部分与dhfr外显子之间以及dhfr外显子与靶基因的3′部分之间发生剪接事件,使得dhfr外显子纳入靶基因mrna中。由于当dhfr外显子被纳入mrna中时选择性dhfr外显子含有框内提前终止密码子,因此降低萤光素酶基因表达。然而,当选择性dhfr外显子的5′ss(即3′内含子的5′末端处的剪接供体位点)发生突变或不可接近从而阻止在此位点处的剪接时,dhfr外显子被从mrna中排除,并且mrna被高效地翻译并且靶基因蛋白质被表达(图2a)。我们首先测试了具有未改变的天然顺式元件的dhfr外显子2以及其中5′ss序列被加强或减弱的各种其他型式的剪接。具有天然5’ss和3’ss的dhfr外显子(seqidno.:8)插入con1中的内含子序列中以形成dhfr_wt与不含选择性dhfr外显子的con1相比显著降低了萤光素酶表达。来自dhfr_wt构建体的表达比con1低116倍(图2b)。当dhfr外显子的5′ss突变为非功能性序列(dhfr_wt5ssc;seqidno.:48)时,dhfr外显子纳入被阻断并且萤光素酶表达恢复到con1的水平(图2aii、2b、2c)。当用更强的5′ss(本情况中为来自con1的5’ss)代替dhfr外显子的5′ss(dhfr_con15ss;seqidno.:49)时,dhfr外显子的纳入增加,从而使得与con1相比萤光素酶基因表达降低545倍(图2b)。然而,当使用来自con4的弱5’ss(dhfr_con45ss;seqidno.:50)时,dhfr外显子未纳入并且萤光素酶表达增加(图2b)。外显子剪接增强子(ese)或抑制子(ess)元件在剪接中起关键作用,并且其功能通常为背景依赖性的。测试了定位于dhfr外显子内的推定剪接调控序列的作用。当定位于dhfr外显子内的推定剪接增强子srp40结合位点发生突变(表2,dhfr_mtsrp40;seqidno.:51)时,dhfr外显子纳入显著增强,使得与con1相比萤光素酶表达降低2982倍(图2c,dhfr_wt和dhfrmtsrp40)。当dhfr外显子内的另一剪接增强子sc35结合位点(通过esefinder预测)突变为更强的sc35结合序列(表2,dhfr_strsc35;seqidno.:52)时,dhfr外显子纳入增强,从而与con1相比使萤光素酶表达降低139倍(图2c,dhfr_wtstrsc35)。这相较于在含有天然dhfr外显子的构建体(dhfr_wt图2b)情况下所观察到的为略微更大的降低。当剪接增强子sc35结合位点突变为剪接抑制子(hnrnpa1结合位点)(表2,dhfr_wtsc35hnrnpa1;seqidno.:53)时,dhfr外显子的纳入效率较低,从而使得萤光素酶表达增加(图2c,dhfr_wt和dhfr_wtsc35hnrnpa1)。已形成了内含子-外显子-内含子盒,其中靶基因(本情况中为萤光素酶)的表达可视选择性外显子(本情况中为含有框内终止密码子的选择性dhfr外显子)的纳入或排除而开启或关闭。引起选择性外显子纳入的剪接使基因表达降低,而当剪接排除选择性外显子时基因表达增加。选择性外显子5’ss的强或弱以及外显子内调节剪接的序列经由其对外源性外显子的纳入或排除的影响来改变目标表达水平。实施例3.选择性外显子5’剪接位点处的发夹形成对调控靶基因表达的影响.实验程序合成(idt)含有具有天然3’和5’ss序列的dhfr外显子2的序列,其中5’ss嵌入发夹结构,并且使用金门克隆策略(neb)将所述序列克隆至指示的载体中。将构建体转染至hek293细胞中并且如实施例1中所描述分析萤光素酶活性。结果我们测试了将dhfr外显子的5’ss嵌入发夹茎部结构是否会影响剪接和选择性dhfr外显子的纳入,并且因此改变靶基因表达(说明于图3a中)。具有con15’剪接位点(dhfr_con15ss;seqidno.:49)序列的选择性dhfr外显子的纳入与con1相比消除萤光素酶表达(图3c,dhfr_con15ss)。将嵌入5′ss的整个序列的15碱基对(bp)发夹结构工程化至dhfr_con15ss中以形成dhfr_con15ss_hp15(seqidno.:54)(图3a)。15bp发夹结构的存在使萤光素酶表达完全恢复到con1的水平,表明发夹已消除5’ss的可接近性,并且从而消除了选择性dhfr外显子的纳入。(图3c,con1,dhfr_con15ss和dhfr_con15ss_hp15)相比之下,具有“被破坏的茎部”的15bp发夹(图3b,con15ss_15hpx;seqidno.:55)不能恢复萤光素酶表达(图3c,dhfr_con15ss_hp15x),表明完整的茎部在调控5剪接位点的可接近性并且从而决定选择性外显子的纳入或排除时是rna二级结构的重要组件。使用含有具有剪接效率增加的突变体srp40结合位点的dhfr外显子的构建体(dhfr_wtmtsrp40,参见实施例2)进行相同的实验。5’ss嵌入发夹使萤光素酶表达恢复,而破坏发夹阻断了萤光素酶表达(图3c,dhfr_wtmtsrp40、dhfr_wtmtsrp40_hp15以及dhfr_wtmtsrp40_hp15x)。因此,将选择性外显子的5’ss嵌入发夹结构可通过阻断所述5′ss的可接近性,并且从而阻止选择性外显子纳入mrna,并且允许靶基因蛋白质表达而使靶基因表达恢复。形成基因表达平台,其中可通过二级rna结构通过改变外源性选择性外显子的5′ss可获得性来调节靶基因蛋白质表达。使用构建体dhfr_wtmtsrp40(seqidno.:58)(以下称为“mtdhfr”)进行进一步核糖开关研发。实施例4.使用茶碱适体通过选择性剪接来调控靶基因表达.实验程序构建dhfr-受体载体以促进附接至具有不同长度的发夹茎部的适体序列的克隆。所用的茶碱适体序列为:ggcgataccagccgaaaggcccttggcagcgtc(seqidno:9)。合成(idt)在5′末端具有4核苷酸突出物的茶碱适体寡核苷酸(“寡核苷酸(oligos)”),退火并连接至bsai消化的dhfr-受体载体。如实施例1中所描述,用具有含有茶碱适体的调控盒的萤光素酶报告基因构建体转染hek293细胞。在转染后四个小时时,抽吸培养基,并且添加含有或不含3mm茶碱的新培养基,并且在茶碱处理之后分析萤光素酶20至24小时。以在存在适体配体的情况下所获得的萤光素酶活性除以在不存在适体配体的情况下所获得的值的商来表示倍数诱导。以占在萤光素酶基因的cds中不含ivs2δ内含子的con1构建体所产生的萤光素酶活性水平(称为最大表达)的百分比来表示萤光素酶活性水平。结果为了调控含终止密码子的选择性外显子的5′剪接位点的可接近性,并且从而调控靶基因蛋白质表达,将适体序列附接至嵌入dhfr5′ss和其互补序列的内含子部分的发夹结构的茎部。在此构型中,适体序列的插入扰乱了发夹茎部的形成,使dhfr5’ss可接近,从而引起选择性dhfr外显子的纳入并且阻止靶基因蛋白质表达(图4a)。当发生适体/配体结合时,如图4b中所描绘,通过配体结合引发的适体构象变化将dhfr5′ss和其互补序列带到一起以得到稳定的茎部形成,从而隐藏dhfr5′ss并且引起dhfr外显子排除和靶基因蛋白质表达。通过将茶碱适体的下部茎部直接连接至发夹茎部(图4c,dhfr_theo1)来测试茶碱适体。如果茎部太长,那么它在不存在适体/配体结合的情况下可形成稳定结构,而如果茎部太短,那么即使在配体存在时它也可能不会形成稳定的茎部。因此,需要优化茎部的长度,使得仅在适体/配体结合时才形成稳定的二级结构。为了确定在存在而不是不存在配体的情况下允许茎部形成的最佳茎部长度,制备许多构建体,其中茶碱适体被克隆至mtdhfr中(描述于实施例2中,表2),并且对茎部进行连续截短。图4c示出了由此连续截短得到的四种构建体。图4d示出了在存在和不存在茶碱的情况下由此缺失系列的构建体产生的萤光素酶表达。在茎部长度从20bp降至9bp的构建体theo1至theo12中,茎部长度足以即使在不存在适体/配体结合的情况下也形成稳定二级结构。因此,在不存在与存在茶碱的情况下均观察到水平类似于con1的萤光素酶表达。在构建体dhfr_theo13情况下,在不存在茶碱的情况下萤光素酶表达被抑制。这指示mtdhfr外显子5’ss的可获得性,从而引起选择性dhfr外显子的纳入和基因表达的抑制。然而,在存在茶碱的情况下,萤光素酶表达开启,相较于不存在茶碱的情况下的表达水平引起43倍诱导,并且占con1对照载体所表达的萤光素酶水平的约56%。因此,产生了哺乳动物开启-核糖开关,其在存在适体配体茶碱的情况下开启靶基因蛋白质表达。实施例5.使用xpt鸟嘌呤适体经由选择性剪接来调控靶基因表达.实验程序使用具有以下序列的xpt-鸟嘌呤适体来构建核糖开关:cactcatataatcgcgtggatatggcacgcaagtttctaccgggcaccgtaaatgtccgactatgggtg(seqidno.:10)。合成(idt)含有鸟嘌呤适体序列和具有4核苷酸5′突出物的发夹茎部的寡核苷酸,退火并且然后连接至bsai消化的dhfr-受体载体。如实施例1中所描述转染hek293细胞。在转染后四个小时时,抽吸培养基,并且添加含有或不含500μm鸟嘌呤的新培养基。在鸟嘌呤处理之后如实施例1和实施例4中所描述分析萤光素酶表达20至24h。使用由atcc推荐的方案培养hepg2、aml12、rd以及c2c12(atcc)。使用吉布森克隆策略(gibsoncloningstrategy)(neb)将含有xpt-g17核糖开关(seqidno.:15)的内含子-外显子-内含子盒插入抗kdr抗体基因的前导肽序列中并且插入小鼠促红细胞生成素基因中的stui位点。将含有小鼠促红细胞生成素(epo)或抗kdr抗体的构建体转染至hek293细胞中。在转染后四个小时时,抽吸培养基,并且添加含有或不含500μm鸟嘌呤的新培养基。将上清液进行elisa分析以产生抗kdr抗体或产生小鼠epo(r&dsystems)。结果通过将衍生自枯草芽孢杆菌(bacillussubtilis)的xpt-鸟嘌呤适体通过茎部p1附接至发夹茎部(图5a,dhfr_g1)来研究使用额外适体/配体通过适体介导的对选择性剪接的调节来控制靶基因表达。类似于实施例4,通过连接茎部的连续截短制备18个构建体(图5a和5b;dhfr-g1至g18,也称为含xpt-g1至g18的调控盒),以获得与鸟嘌呤适体连接的发夹茎部的最佳长度,从而允许适体/配体结合与5’ss可接近性和外显子剪接相联系。如图5b中所示,在茎部长度从24bp降至12bp的构建体dhfr-g1至g13的情况下,在存在或不存在适体配体鸟嘌呤的情况下萤光素酶表达不受选择性dhfr外显子的插入和xpt-鸟嘌呤适体的影响。这表明茎部的长度足以在不存在与存在配体的情况下均形成稳定的结构,从而阻止选择性外显子纳入mrna。然而,在构建体dhfr_g14至dhfr_g18中,在不存在所添加的鸟嘌呤的情况下萤光素酶表达被抑制。当添加μm鸟嘌呤时,来自这些构建体的萤光素酶表达被诱导(图5b)。在鸟嘌呤处理后,对构建体g11至g18的进一步严格验证再次证实对萤光素酶表达的明显调控(图5c)。含有xpt-g17的构建体dhfr_g17(seqidno.:15)(图5a)给出2000倍表达诱导,产生由con1表达的萤光素酶水平(称为最大表达)的约65%。相较于在不存在配体的情况下极低的未诱导基线水平,此高动态范围的诱导是由表达的激活引起的。构建体dhfr_g16(图5a)相较于未诱导基线表达给出约800倍的诱导,达到为最大表达的83%的水平(图5c和5d)。此外,构建体dhfr_g14和g15展示近100%的最大表达,倍数诱导较低,这是归因于萤光素酶的未诱导基线表达较高。为测试合成核糖开关在内含子-外显子-内含子盒中的一般功能和适用性,我们将含xpt-g17构建体(dhfr_g17)转染至多个人类和小鼠细胞系中。在这些不同细胞系中,鸟嘌呤处理产生显著的基因表达诱导,在hepg2细胞中为超过500倍诱导,在其他细胞系中倍数诱导较低(图5e)。不同细胞系中的不同倍数诱导可反映在转染效率方面以及在细胞类型特异性剪接调控剂表达型态方面的差异。此外,具有含有xpt-g17核糖开关的调控盒的萤光素酶基因(dhfr_g17)当转移至aav骨架时产生相似水平的诱导(图5f),表明基因调控作用不是载体骨架依赖性的。除了调控萤光素酶基因,还在调控所分泌蛋白质、抗kdr抗体以及促红细胞生成素(epo)方面对含xpt-g17调控盒进行了测试。将含xpt-g17调控盒插入抗kdr抗体和促红细胞生成素的编码序列中。如图5g和5h中所示,当与在不存在配体的情况下由细胞产生各分子相比时,鸟嘌呤处理在抗kdr抗体制备中产生80倍诱导,并且在epo制备中产生140倍诱导。这些结果证实了潜在治疗性靶基因在调控蛋白质表达中的一般功能和适用性,以及此基因调控盒在aav介导的基因递送中的应用。因此,我们形成了合成的哺乳动物“开启”-核糖开关,其能够响应于哺乳动物细胞中适体特异性配体的存在而开启靶基因蛋白质表达。实施例6.可使用不同嘌呤适体经由选择性剪接来调控靶基因表达.实验程序使用以下在表3中所列的适体序列来构建核糖开关:表3.结果为在我们的基因调控系统中测试额外的适体,我们使用与先前实施例中所描述相同的策略和方法通过将不同的鸟嘌呤和腺嘌呤适体连接至内含子-mtdhfr-内含子盒来产生多个鸟嘌呤和腺嘌呤响应性核糖开关(图6a)。所测试的鸟嘌呤核糖开关响应于鸟嘌呤而高效地调控萤光素酶基因的表达(图6b)。另外,我们发现这些鸟嘌呤核糖开关不仅响应于鸟嘌呤(图6b)而且还响应于鸟苷(图6c)以及2’脱氧鸟苷而调控靶基因的表达(2’dg)(图6d)。产生了许多腺嘌呤核糖开关(图6a),并且也展示基因调控功能(图6e)。所测试的含有不同适体的构建体在调控基因表达方面的差异可反映适体/配体结合亲和力和适体二级结构的差异,这些差异可能会影响5′ss的可接近性、选择性外显子纳入并且因此影响靶基因表达。可通过改变适体序列对内含子-外显子-内含子基因调控盒进行优化,以实现所需基因调控水平。实施例7.由哺乳动物鸟嘌呤核糖开关调控的靶基因表达的开启/关闭状态.实验程序对内含子-mtdhfr-适体-内含子盒进行pcr扩增并且使用金门克隆策略(neb)克隆至pegfp-c1载体中。为获得使用核糖开关稳定表达egfp的细胞系,用20ng质粒dna将hek-293细胞电穿孔。电穿孔后四八小时时,用800μg/ml的g418对细胞培养物进行选择持续2周以获得稳定表达所述盒的细胞。对细胞进行胰蛋白酶消化,并且使用guavaeasycyte8ht机器对细胞悬浮液进行gfp荧光强度的流式细胞分析。使用guavasoft2.2.2分析所得数据。结果为进一步证实可通过暴露于对核糖开关内所含的适体具特异性的配体来调控含有我们的内含子-外显子-内含子调控盒的靶基因的表达,将含有xpt-g17核糖开关(seqidno.:15)的内含子-mtdhfr-内含子盒插入egfp基因中,并且稳定转染hek293细胞。在存在鸟嘌呤的情况下,egfp表达开启(图7a)。早在鸟嘌呤处理后6小时时就检测到荧光,并且历经3天的鸟嘌呤处理而增加,与未处理的细胞相比达到接近300倍诱导(图7b),指示在存在适体配体的情况下靶基因表达的“开启”状态。当鸟嘌呤从细胞培养基中撤出时,egfp表达减少,指示在不存在适体特异性配体的情况下靶基因表达的“关闭”状态(图7b)。因此,我们已形成了一种基因调控平台,所述基因调控平台包含含有合成核糖开关的内含子-外显子-内含子盒,通过所述合成核糖开关在哺乳动物细胞中通过存在或不存在特异性适体配体而对靶基因的表达进行调控。实施例8.多个调控盒对调控靶基因表达的影响.实验程序使用金门克隆策略(neb)制备构建体。将hek293细胞用所指示的构建体转染,在转染后用500μm鸟嘌呤或1mm鸟苷(sigma)处理4h。如实施例5中所描述分析萤光素酶活性。结果具有含有xpt-g15(seqidno.:46)的调控盒的构建体(dhfr_g15;实施例5)响应于鸟嘌呤处理与未诱导基础表达水平相比展示60倍萤光素酶表达诱导,并且达到由con1表达的萤光素酶水平的近100%(图8a)。当需要高水平地调控治疗性蛋白质时,这是一个有用的特征。相比之下,具有含xpt-g17调控盒的构建体(dhfr_g17)具有2181倍的显著更高的倍数诱导,这是归因于未诱导基线表达较低,但与con1相比在诱导后最大表达水平明显更低(图8a)。为测试两个拷贝的含xpt-g15调控盒(xpt-g15双;seqidno.:64)是否可降低基础表达水平,而不损害在诱导后萤光素酶的最大表达水平,将两个拷贝的含xpt-g15调控盒嵌入萤光素酶基因中,各拷贝在基因序列中位于不同位置。当存在两个拷贝的含xpt-g15调控盒时,未诱导基线表达降低,从而产生显著更高的诱导倍数(60倍至1008倍),而不损害最大表达水平(图8a)。xpt-g15双盒(xpt-g15双;seqidno.:64)的鸟嘌呤ec50比含有单个拷贝的更严格的含xpt-g17盒的构建体的鸟嘌呤ec50低5倍(43μm相较于206μm),从而增加配体反应的敏感性(图8a)。使用两个拷贝的较不严格的调控盒来增强倍数诱导和最大诱导基因表达的策略也适用于egfp基因。如图8b和8c中所示,与萤光素酶调控的结果(图8a)一致,当与含xpt-g17调控盒的构建体(egfp-xpt-g17)相比较时,含单一拷贝的xpt-g15调控盒的egfp基因(egfp-xpt-g15)产生较高的未诱导基线egfp表达水平。然而,当将两个拷贝的xpt-g15调控盒在不同位置插入egfp基因时(egfp-xpt-g15双),未诱导基线表达水平降低至egfp-xpt-g17的未诱导基线表达水平,并且诱导egfp水平甚至高于con1-egfp对照(图8c)。此外,将一个拷贝的含xpt-g17调控盒和一个拷贝的含有ydhl-a5腺嘌呤核糖开关的调控盒嵌入萤光素酶基因中。通过单独添加腺嘌呤(25倍)或添加鸟嘌呤(120倍)来诱导萤光素酶表达,然而,在以各自使用的最高浓度组合使用腺嘌呤和鸟嘌呤的情况下实现显著更高水平的诱导(高达2966倍)(图8d).这些结果证实基于选择性剪接的核糖开关在调控靶基因表达中的模块性。为了减少重组并且增加产生含有两个或更多个调控盒的病毒载体的容易性,可在单个靶基因中使用具有不同内含子和外显子序列的调控盒,并且这些调控盒可含有相同或不同的配体响应性适体。实施例9.内含子尺寸和序列对经由适体介导的选择性剪接调控靶基因表达的影响.实验程序使用con1构建体作为内含子片段的pcr扩增模板,所述内含子片段具有上游或下游内含子缺失。为产生具有单一内含子缺失的构建体,使用金门克隆策略(neb)将pcr产物克隆至含有xpt-g17核糖开关的构建体中。为产生具有上游与下游内含子缺失的构建体,将由ecori和bamhi从在下游内含子序列中具有单一缺失的构建体释放的片段克隆至ecori和bamhi消化的在上游内含子序列中具有单一缺失的构建体中。结果内含子含有可能会促进(内含子剪接增强子,ise)或抑制(内含子剪接抑制子,iss)外显子剪接的元件。在我们已产生的所有核糖开关中,xpt-g17在诱导倍数与诱导基因表达水平方面均展示最佳调控能力。使用内含子-外显子-内含子盒中的xpt-g17核糖开关,我们在内含子序列和内含子长度以及剪接位点中进行了一系列修饰以进一步优化系统。首先,通过在内含子序列中在mtdhfr外显子的上游或下游(图9a、9b)引入单一缺失测试了内含子修饰的影响,产生了16个具有含xpt-g17核糖开关的构建体xpt-g17-ir-1至xpt-g17-ir-16(这些构建体中有13个的序列在表5中给出,seqidno:16-seqidno:28)。然后,如图9c中所描绘,将上游和下游内含子缺失组合以产生更大的内含子缺失。如图9d和9e中所示,制备具有两处内含子缺失(2ir)的16个构建体,构建体2ir-1至2ir-10(seqidno.:29-seqidno.:38)展示显著更高的诱导倍数,而不损害诱导萤光素酶表达水平,其中2ir-3的倍数诱导具有最大的改善(4744倍)。此外,我们还制备了在mtdhfr外显子的上游具有突变的3′ss的构建体,并且还减小了下游内含子的尺寸。如图9d和9e中所示(构建体dhfr_3ssc_1至5),这些修饰进一步改善了相对倍数诱导,然后,在此情况下观测到诱导表达水平的降低(对于3ssc_3来说从64%至32%)。这些结果表明,可通过对侧接选择性外显子的内含子序列进行修饰来优化内含子-外显子-适体-内含子盒的基因调控能力以实现所需基因调控水平。实施例10.在基因调控盒中使用多个天然外显予以及合成外显子.实验程序合成(idt)突变体人类维尔姆斯瘤1外显子5(mutwt1-e5,seqidno.:61)、sirt1外显子6(sirt1-e6,seqidno.:62)、小鼠钙/钙调蛋白依赖性蛋白激酶iiδ外显子16或17(camk2d-e16或e17,seqidno.:59、seqidno.:60)以及合成外显子eneee(seqidno.:63)的序列,并且将其代替dhfr外显子使用吉布森克隆试剂盒(neb)克隆至dhfr-g17载体中。将质粒dna转染至hek293细胞中,用500μm鸟嘌呤处理,并且如实施例1中所描述进行萤光素酶分析。示出了具有5′和3′剪接位点的各外显子的序列,外显子序列以大写字母显示(表5,seqidno.:59至seqidno.:63)。结果为了确定四个内含子-外显子-适体-内含子盒的调控功能不限于特定的外显子序列,我们将含有鸟嘌呤xpt-g17核糖开关的构建体(dhfr-xpt-g17)中的mtdhfr外显子用含有已知外显子剪接增强子序列(ese)的多个不同的天然和突变体外显子以及合成外显子代替。如图10中所示,具有camkiid-e16外显子的调控盒与dhfr-xpt-g17相比产生几乎等效的倍数诱导,但基础与诱导萤光素酶表达的水平均较低。含有其他外显子的盒也展示可变水平的基础与诱导萤光素酶表达。因此,适体介导的选择性剪接基因调控盒不是外显子特异性的,并且不限于mdhfr-e2外显子。可产生高效选择性剪接事件的外显子适用于适体介导的基因调控盒。这些结果进一步表明,可通过对选择性外显子中的序列以及周围内含子序列,例如如本文中所描述的选择性外显子的5’ss和3’ss序列的剪接强度以及选择性外显子中的ese和ess序列进行修饰来优化此内含子-外显子-适体-内含子盒的基因调控能力。实施例11.在小鼠中在体内通过适体介导的选择性剪接来调控靶基因表达.实验程序水动力dna递送和药物处理:通过尾部静脉以10%体重的体积历时5至10秒向6-7周龄cd-1雌性小鼠注射5μg或10μg的在盐水中稀释过的不含内毒素的质粒dna(qiagen无内毒素试剂盒),所述质粒dna含有具有两个拷贝的含xpt-g15调控盒的萤光素酶基因(xpt-g15双,seqidno.:64;实施例8,图8a)。将鸟苷(sigma)新鲜悬浮于含0.5%甲基纤维素/0.25%吐温80(tween80)(sigma)的水中,并且在dna注射后2h和12h时经口施用,或在dna递送后5h、12h、16h以及24h时通过腹膜内注射(ip)进行递送。无创活体动物生物发光成像:在成像之前,将小鼠用2%异氟烷麻醉,并且注射每千克体积150gm荧光素,并且在dna注射后的所指示时间点使用brukerxtreme系统在荧光素注射后的2至5分钟内获取图像。以平均光子/秒±标准偏差来表示萤光素酶活性。以在用鸟苷处理的小鼠中所获得的平均光子/秒除以在未进行鸟苷处理的小鼠中所获得的值的商的形式来计算诱导倍数。结果我们评估了内含子-外显子-内含子调控盒在小鼠中在体内中的基因调控功能。通过流体力学注射将在萤光素酶基因中含有两个拷贝的xpt-g15核糖开关的构建体(xpt-g15双)的不含内毒素的质粒dna递送至小鼠的肝脏,并且腹膜内施用鸟苷。我们测试了两种鸟苷递送途径。在一个实验中(图11a和11b),在dna递送后2h和12h时经口为小鼠施用不同剂量的鸟苷,然后成像。如图11a中所示,用鸟苷处理的小鼠在dna后9小时时展示较高的萤光素酶表达。在鸟苷处理的小鼠中萤光素酶表达随时间推移而增加,在dna注射后48小时时达到最高水平,此后表达下降。在单独的实验中(图11c和图11d),腹膜内施用鸟苷。在dna注射后(p.i.)4小时时,并且在鸟苷处理后,各组中的小鼠展示出类似水平的基础萤光素酶活性(图11)。然后,用媒介物处理小鼠作为对照或者用鸟苷处理。在dna注射后11小时时,在所有小鼠中萤光素酶活性增加,这与在肝脏中在水动力dna注射后12小时时萤光素酶基因表达达到峰值的报道相一致。然而,与未处理的小鼠中相比,在用鸟苷处理的小鼠中,存在显著更高水平的萤光素酶表达,与未诱导基线表达相比在100mg/kg和300mg/kg鸟苷的情况下分别观察到4.7倍和16.2倍诱导。因此,证实基于剪接的基因调控盒在动物中在体内响应于对调控盒内所含的适体具特异性的配体的施用以剂量依赖性方式调控基因表达。实施例12.经由腺相关病毒(aav)载体将核糖开关构建体递送至鼠类视网膜.实验程序aav质粒构建体:经由分子克隆成能够被包装为aav基因组的形式来使两个核糖开关表达构建体(描述于下表中)适应。表4.将基于egfp转基因的表达构建体(gtx5-7)用限制酶mfei和nhei消化,释放约1400bpdna片段,所述dna片段含有核糖开关诱导元件和egfp转基因。还将pd10aav基因组质粒用mfei和nhei消化,释放4475bp片段,所述片段含有aavitr、cmv启动子以及sv40聚腺苷酸化信号。使用t4dna连接酶连接两个片段,产生质粒,所述质粒含有具有以下结构的序列,能够被包装为aav2基因组:[itr]-[cmv]-[5’egfp]-[核糖开关元件]-[3’egfp]-[sv40]-[itr]通过dna测序验证所有所得质粒构建体,并且根据以下惯例来命名:pd10-gtx#。aav载体制备和滴定:在体外通过用三种质粒瞬时转染hek-293t细胞来产生腺相关病毒(aav)。(i)基于pd10骨架的病毒基因组质粒(ii)含有aav2rep78基因和病毒衣壳基因的aav包装质粒。可通过改变衣壳基因序列产生aav的许多不同血清型,本情况中使用aav8衣壳。(iii)辅助质粒(phgti-adeno1)。此质粒提供aav需要包装和组装的接近最小的腺病毒基因集合。以比率1∶1∶3将这些质粒转染至hek-293t细胞中,每一80-90%汇合的150cm2板转染总计50μg的质粒dna。典型的制备操作由20个这样的板组成。所用的转染试剂为聚乙烯亚胺(pei),pei与dna比率为2.25∶1(w/w)。转染后七十二小时时,使细胞从板上物理脱离并且通过离心形成沉淀;将所得细胞沉淀再悬浮于20ml的tris密度缓冲液中。然后通过反复冷冻/解冻/涡旋循环将沉淀裂解,并且通过核酸酶消化将残留在裂解物中的任何未包装dna破坏。然后通过死端过滤和离心使裂解物澄清,然后稀释至总体积为50ml。然后经由基于亲和力的fplc程序在根据预先编程的方案运行的aktapure仪器上使用avb柱(均来自gehealthcare)将澄清的裂解物纯化。通过在10,000mw截止值的vivaspin4离心浓缩机(gehealthcare)中在5000xg下离心将含有来自fplc柱的洗脱液的最终aav浓缩降至约200μl的体积,添加2ml的pbs-mk(以稀释出高盐洗脱缓冲液),并且使用同一浓缩机将洗脱液再浓缩回约200μl。此材料构成了纯化的aav病毒,并且被适当等分并储存在-80℃下。使用直接对纯化载体的样品进行qpcr(针对sv40聚腺苷酸化信号)来确定载体滴度。将所得循环阈值与已知标准曲线进行比较,并且计算每毫升的载体基因组的数目。根据以下惯例对核糖开关aav载体进行命名:aav2/[衣壳血清型#]-gtx#鼠类视网膜下注射:在全身麻醉下,使用安装在5μl汉密尔顿注射器(hamiltonsyringe)上的手动引导的10mm34号针头对小鼠进行注射,将载体注射至视网膜下空间。通过经由操作显微镜观察视网膜将针尖引导至注射位置。在所有接受载体的眼睛中,进行2次2μl注射,一次注射位于眼睛的上半球,而另一次注射在下半球。在注射后,记录所得视网膜脱离的质量和注射材料的任何回流。荧光眼底照像:在视网膜下注射之后,使用附带有leicadc500数码相机的裂隙灯(sc-16,keeler)通过眼底照像定期评估egfp转基因表达。将动物置于全身麻醉下,并且用1%局部托吡卡胺(tropicamide)扩张其瞳孔。通过将盖玻片放置于用耦合介质溶液(viscotears)覆盖的角膜上来中和角膜屈光力。在明亮的白光下,调整仪器,并且安置动物,使得视网膜处于锐聚焦状态,并且视盘在视野中央,然后使用200ms曝光时间获取明视野图像。通过过滤光源(475±25nm)并且以10和30s曝光再获取两个图像来评估转基因(egfp)荧光。结果如通过连接产物的dna测序所示,将核糖开关构建体(表4)成功克隆成能够被包装成aav基因组的形式。在所得构建体中,将pd10-gtx7和pd10-gtx5进一步制备成aav2/8病毒载体。通过qpcr证实所制备的载体具有以下滴度:aav2/8-gtx7:1.17x1013个载体基因组/mlaav2/8-gtx5:1.73x1013个载体基因组/ml然后视网膜下注射这两种载体,并且放置8天以进行egfp转基因表达,然后通过荧光眼底照像来评估表达。图12证实egfp在注射有aav2/8-gtx7的视网膜中被表达。转基因表达较低,但将仅在响应于配体(未添加)经由适体介导的选择性剪接的诱导之后预期aav2/8-gtx7的基本表达。实施例13.在鼠类视网膜中在aav递送后在体内通过调控盒介导的选择性剪接来调控靶基因表达.程序荧光眼底照像(egfp信号)的定量:使用gnu图像操作程序(gimp,开放来源)进行对图像的所有操作和分析。如上文所描述,在各成像点获取各视网膜的三个图像:白光(200ms)、475±25nm(10s)以及475±25nm(30s)。首先将这三个图像以层的形式叠加,并且使用白光图像作为指导,将感兴趣的区域(roi)定义成涵盖通过瞳孔可见的整个视网膜。对两个475±25nm(egfp荧光)图像使用阈值工具,仅突出显示强度值超过所定义的阈值的那些像素。在确定egfp信号与背景清楚分离的基础上选择阈值,并且为分析提供适当的动态范围。记录每个图像在roi内超过阈值的像素的数目。为了校正导致眼睛之间的可见视网膜区域变化的可变瞳孔扩张,将超过阈值的像素的数目除以roi内的像素总数目。诱导:经由如下文所描述的两种施用途径进行核糖开关介导的对靶基因表达的诱导:腹膜内注射(i.p.):使用13mm30号针头将100μl体积的[75mg/ml鸟苷+含0.5%w/v甲基纤维素+0.25%v/v吐温80的水]注射至腹膜内空腔中。这相当于在体重25g的成年小鼠中鸟苷剂量为300mg/kg。玻璃体内注射(i-vit.):使用手动引导的10mm34号针头玻璃体内注射2μl体积的[1mm鸟苷+含2.5%dmso的pbs-mk]。注射时的针尖位置为晶状体下方,视盘正上方,通过经由操作显微镜观察视网膜已将其引导至此位置。结果在第00天时,如实施例12中所描述,如下视网膜下注射总共9个眼睛:-6个眼睛使用aav2/8-gtx7(通过g15核糖开关元件调控的由cmv启动子引起的egfp转基因表达)-3个眼睛使用aav2/8-gtx5(阳性对照构建体,未调控的由cmv启动子引起的egfp转基因表达)在第02、08、09、10以及12天如实施例12中所描述进行荧光眼底照像。在第08、09以及10天在荧光眼底照像之后所有眼睛经由腹膜内注射接受诱导。在第11天所有眼睛经由玻璃体内注射接受诱导。如上文所描述对荧光信号进行定量,并且将示例性图像在图13a中示出。在载体注射后头8天期间不进行诱导,因为已知由aav2/8引起的基因表达需要长达7天才能变为最大值。因此,将第8天的表达水平作为诱导前基线。如图13c和图13a(l相较于n)中所示,在载体注射后第10天在2轮腹膜内诱导之后,与此基线相比转基因表达已增加了约3.5倍(p≤0.05,单因素方差分析,唐那氏)。如图13c和图13a(l相较于o)中所示,在载体注射后第12天,在玻璃体内诱导之后24h以及在最后一次腹膜内诱导之后48h时,与基线相比转基因表达已增加了约9倍(p≤0.001,单因素方差分析,唐那氏)。在玻璃体内诱导之后此大得多的诱导暗示(但并不明确地表明)此诱导途径可能比腹膜内注射更有效。图13b中示出了显示诱导之前和之后egfp转基因表达的差异的更高分辨率图像。历经相同的时间段并且在相同诱导方案下,由未调控的对照载体aav2/8-gtx5介导的egfp表达水平不显著改变(单因素方差分析,邦弗朗尼),如图13d中所示保持大致恒定。由于gtx7与gtx5介导的表达水平的差异较大,各图像集合需要不同的曝光时间(分别为30s和10s)和阈值(分别为50和190)。该数据清楚地表明,由基于g15的gtx7构建体引起的转基因表达是经由鼠类视网膜中的适体介导的选择性剪接来调控。由gtx7引起的诱导的最大转基因表达水平低于由不可诱导的阳性对照构建体gtx5介导的最大转基因表达水平。表5.描述和相关序列。除非另有说明,否则外显子序列以大写字母显示,而内含子序列以小写字母显示。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1