π共轭类化合物、有机电致发光元件材料、发光材料、发光性薄膜、有机电致发光元件、显示装置及照明装置的制作方法
本发明涉及一种π共轭类化合物、有机电致发光元件材料、发光材料、发光性薄膜、有机电致发光元件、显示装置及照明装置。
背景技术:
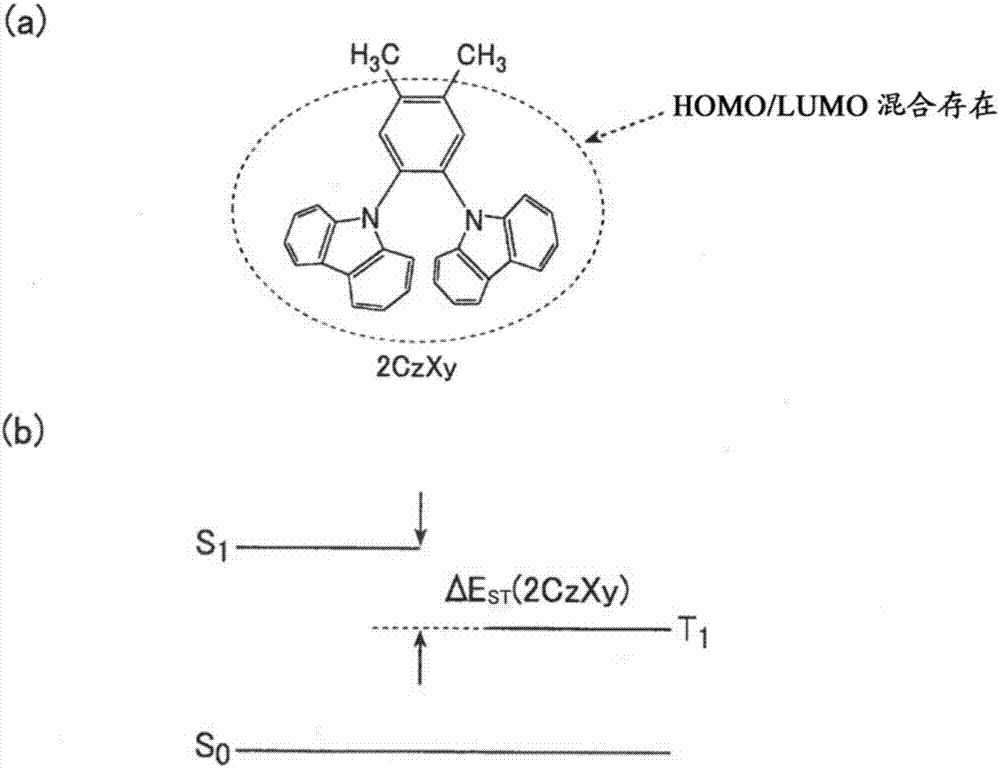
:利用了有机材料的电致发光(electroluminescence:以下简称为“el”。)的有机电致发光元件(也称为“有机电场发光元件”、“有机el元件”)为是可以作为进行平面发光的新发光系统已经被实用化的技术。有机el元件从基本的电子显示器开始,最近也被适用于照明设备,并期待其发展。作为有机el元件的发光方式,包括在从三重态激发状态返回到基态时发出光的“磷光发光”、和在从单态激发状态返回到基态时发出光的“荧光发光”这两种。对有机el元件施加电场时,从阳极和阴极分别注入空穴和电子,在发光层中进行再键合而产生激发子。此时,单态激发子和三重态激发子以25%:75%的比例生成,因此,已知利用三重态激发子的磷光发光与荧光发光相比,理论上可得到高的内部量子效率。但是,为了在磷光发光方式中实际上得到高的量子效率,有必要采用中心金属使用铱或铂等稀有金属的络合物,担心将来稀有金属的埋藏量和金属本身的价格成为产业上大的问题。另一方面,为了在荧光发光型中提高发光效率,进行了各种开发,近年来出现新的动态。例如,在专利文献1中,公开如下技术:通过两个的三重态激发子的冲突而生成单态激发子的现象(triplet-tripletannihilation:以下,适当简称为“tta”。另外,着眼于triplet-tripletfusion:也称为“ttf”),有效地引起tta而实现荧光元件的高效率化。利用该技术,荧光发光材料的发光效率提高至以往的荧光发光材料的2~3倍,但tta中的理论的单态激发子生成效率停留在40%左右,因此,依然与磷光发光相比,具有高发光效率化的问题。另外,近年来,报道了下述荧光发光材料和将其利用于有机el元件的可能性,所述荧光发光材料利用了下述现象(热活性型延迟荧光(也称为“热激发型延迟荧光”:thermallyactivateddelayedfluorescence:以下,适当简称为“tadf”),所述现象利用了产生从三重态激发子向单态激发子的逆系间穿越(reverseintersystemcrossing:以下,适当简称为“risc”。)的现象(例如参照专利文献2、非专利文献1、非专利文献2)。利用该tadf机制引起的延迟荧光时,在电场激发引起的荧光发光中,也可以产生理论上与磷光发光同等的100%的内部量子效率。为了出现tadf现象,必须引起从在室温或发光元件中的发光层温度下通过电场激发而产生的75%的三重态激发子向单态激发子的逆系间穿越。另外,由逆系间穿越而产生的单态激发子与通过直接激发而产生的25%的单态激发子同样地进行荧光发光,由此,理论上可以产生100%的内部量子效率。为了引起该逆系间穿越,要求最低激发单态能级(s1)和最低三重态激发能级(t1)之差的绝对值(δest)小。例如为了显现tadf现象,缩小有机化合物的δest是有效的,为了缩小δest,分子内的最高被占分子轨道(homo)和最低空分子轨道(lumo)不混合存在而是使其集中分布(明确地发生分离)是有效的。另外,已知在由主体材料和发光材料构成的发光层中含有显示tadf性的材料作为第三成分(辅助掺杂剂材料)时,对高发光效率的显现是有效的(参照非专利文献3)。通过在辅助掺杂剂材料上通过电场激发产生25%的单态激发子和75%的三重态激发子,三重态激发子可以伴随逆系间穿越(risc)而生成单态激发子。单态激发子的能量可以向发光材料进行荧光共振能量移动(fluorescenceresonanceenergytransfer:以下,适当简称为“fret”),通过发光材料移动来的能量而进行发光。因此,可以理论上利用100%的激发子能量使发光材料发光,显现高发光效率。图1及图2是表示显现tadf现象的化合物(tadf化合物)和一般的荧光发光材料的能级图的示意图。例如,在具有图1所示的结构的2czpn中,homo集中存在于苯环上的1位和2位的咔唑基上,lumo集中存在于4位和5位的氰基上。因此,可以将2czpn的homo和lumo进行分离,δest变得非常小而显现tadf现象。另一方面,将2czpn的4位和5位的氰基取代为甲基的2czxy(图2)中,结构类似,但不能进行2czpn中所看到的homo和lumo的明确分离,因此,不能缩小δest,不能达到显现tadf现象。在现有技术中,为了将homo和lumo明确地进行分离,已知有使用强力的供电子基团或吸电子基团的方法。但是,由于使用强力的供电子基团或吸电子基团形成强的分子内电荷移动(ct)性的激发状态,因此,成为吸收光谱或发光光谱发生长波长化的主要原因,产生难以控制发光波长技术问题。相反,为了控制发光波长,减弱供电子性或吸电子性时,导致在表现出tadf现象中出现障碍。因此,期望一边控制发光波长,一边显现tadf现象的新型的方法。但是,近年来,还要求提高有机el元件的生产稳定性或品质稳定性(可靠性)。有机el元件通常具有阴极、阳极、和配置于它们之间的多个有机层。具体而言,有机el元件依次具有阴极/电子注入层/电子传输层/发光层/空穴传输层/空穴注入层/阳极。有机层的数多时,相应地制造工序数也变多,因此,生产稳定性容易损坏。因此,在提高有机el元件的生产稳定性的观点上,要求减少阴极和阳极之间的有机层的数,优选设为仅发光层。另外,发光层通常含有发光性化合物和主体化合物的2种以上。发光层的构成成分的种类多时,需要调整这些构成成分的浓度,同时进行蒸镀,生产稳定性或品质稳定性容易损坏。因此,在提高有机el装置的生产稳定性或品质稳定性(可靠性)的观点上,要求尽可能减少发光层的构成成分的种类,优选不含有主体化合物,可以仅由发光性化合物构成。即,从提高有机el元件的生产稳定性或品质稳定性(可靠性)的观点出发,要求1)减少阴极和阳极之间的有机层的层数、或2)减少发光层的构成成分的种类,使有机el元件的构成尽可能简单化。与此相对,正在研究在发光层中以高浓度含有荧光发光性化合物或磷光发光性化合物的方法。但是,在发光层中以高浓度含有荧光发光性化合物或磷光发光性化合物时,存在元件的发光效率降低的问题。因此,这些化合物不能单独构成发光层,需要还含有主体化合物。还提出了在发光层中不含有主体化合物而以高浓度含有发光的tadf化合物的方法(例如参照非专利文献4)。现有技术文献专利文献专利文献1:国际公开第2010/134350号专利文献2:日本特开2013-116975号公报非专利文献非专利文献1:h.uoyama,etal.,nature,2012,492,234-238非专利文献2:q.zhangetal.,nature,photonics,2014,8,326-332非专利文献3:h.nakanоtani,etal.,naturecommunicaion,2014,5,4016-4022.非专利文献4:advancedmaterialsdoi:10.1002/adma.201405474技术实现要素:发明所要解决的技术问题但是,非专利文献4所示的具有发光的tadf化合物单独构成的发光层的有机el元件均在连续驱动时的耐久性低,实用上不具有充分的功能。作为劣化快的主要原因,认为是tadf化合物中的二苯基磺酰基的分解(参照journalofphysicalchemistryc2014,118,7569-7578)。另一方面,即使非专利文献1或3所示的4czipn或2czpn等tadf化合物单独构成,也不能使其发光,需要还含有主体化合物。因此,并非充分且简单地形成有机el元件的构成。本发明是鉴于上述问题、状况而完成的发明,其解决的技术问题在于,提供一种例如可以改良有机电致发光元件的发光效率的新型π共轭类化合物。另外,提供一种使用了该π共轭类化合物的有机电致发光元件材料、发光材料、发光性薄膜等。另外,提供一种有机电致发光元件、及具备该有机电致发光元件的显示装置及照明装置,所述有机电致发光元件可以不使发光效率和耐久性降低而使有机电致发光元件的构成简单,生产率稳定性及可靠性高。用于解决技术问题的手段本发明人为了解决上述技术问题,对上述问题的原因等进行了研究,结果发现,重新想到一种有机电致发光元件,所述有机电致发光元件的特征在于,含有将苯环的邻位的至少一个组合用供电子部之间的组合、吸电子部之间的组合、或供电子部和吸电子部的组合中任一种组合取代而得到的π共轭类化合物,可以提高发光效率,直至本发明。即,本发明的上述技术问题通过以下的技术方案来解决。[1]一种π共轭类化合物,其具有下述通式(1)所表示的结构。[化学式1](通式(1)中,z1~z6分别表示氢原子、氘原子、供电子基团d或吸电子基团a,z1~z6中的至少两个中的一个为所述供电子基团d,另一个为所述吸电子基团a,并且,z1~z6中的邻位组合z1和z2、z2和z3、z3和z4、z4和z5、z5和z6及z6和z1的至少一个为所述供电子基团d和所述供电子基团d、所述吸电子基团a和所述吸电子基团a、或所述供电子基团d和所述吸电子基团a的组合中的任一种,所述供电子基团d为选自被供电子基团取代的芳基、任选被取代的供电子性杂环基、任选被取代的氨基、及烷基中的基团,所述吸电子基团a为选自氟原子、被氟原子取代的烷基、任选被取代的羰基、任选被取代的硫酰基、任选被取代的氧化膦基、任选被取代的硼烷基、任选被吸电子基团取代的芳基、以及任选被取代的吸电子性的杂环基构成的组中的基团)。[2]如[1]所述的π共轭类化合物,其用下述通式(2)~(10)或(101)的任一种表示。[化学式2](通式(2)~(10)或(101)中,d1~d5分别独立地表示所述供电子基团d;z1~z5分别独立地表示氢原子、氘原子或所述吸电子基团a。)[3]如[1]或[2]所述的π共轭类化合物,其用下述通式(11)~(42)的任一种表示。[化学式3](通式(11)~(42)中,d1~d5分别独立地表示所述供电子基团d;a1~a5分别独立地表示所述吸电子基团a。)[4]如[3]所述的π共轭类化合物,其中,所述通式(11)~(42)中,d1~d5分别独立地包含选自被供电子基团取代的苯基、任选被取代的咔唑基、任选被取代的氮杂咔唑基、任选被取代的二氮杂咔唑基、任选被取代的9,10-二氢吖啶基、任选被取代的吩噁嗪基、任选被取代的吩噻嗪基、任选被取代的5,10-二氢吩嗪基、任选被取代的二苯基氨基、任选被取代的二烷基氨基中的基团构成,且a1~a5分别独立地包含选自被氰基取代的芳基、未取代的含氮芳香族6元环基、被氟原子取代的含氮芳香族6元环基、被氰基取代的含氮芳香族6元环基、被氟取代烷基取代的含氮芳香族6元环基、被任选被取代的芳基取代的含氮芳香族6元环基中的基团。[5]如[3]或[4]所述的π共轭类化合物,其用所述通式(11)~(15)、(18)、(19)、(23)、(24)、(26)、(27)、(29)、(30)、(39)、(40)或(42)表示。[6]如[3]~[5]中任一项所述的π共轭类化合物,其用所述通式(13)、(15)、(18)、(19)、(23)、(24)、(27)、(30)、(40)、或(42)表示。[7]如[3]~[6]中任一项所述的π共轭类化合物,其中,所述通式(11)~(42)中,满足d1=d2=d3=d4=d5和a1=a2=a3=a4=a5中的至少一个。[8]如[1]~[7]中任一项所述的π共轭类化合物,其中,最低激发单态能级和最低激发三重态能级的能量差的绝对值δest为0.50ev以下。[9]一种有机电致发光元件材料,其含有[1]~[8]中任一项所述的π共轭类化合物。[10]一种发光材料,其含有[1]~[8]中任一项所述的π共轭类化合物,所述π共轭类化合物放射荧光。[11]如[10]所述的发光材料,其中,所述π共轭类化合物放射延迟荧光。[12]一种发光性薄膜,其含有[1]~[8]中任一项所述的π共轭类化合物。[13]如[12]所述的发光性薄膜,其中,所述π共轭类化合物放射延迟荧光。[14]一种有机电致发光元件,其含有阳极、阴极、及设置于所述阳极和所述阴极之间的发光层,所述发光层的至少一层含有[1]~[8]中任一项所述的π共轭类化合物。[15]如[14]所述的有机电致发光元件,其中,所述π共轭类化合物生成激发子。[16]如[14]或[15]所述的有机电致发光元件,其中,所述π共轭类化合物放射荧光。[17]如[16]所述的有机电致发光元件,其中,所述π共轭类化合物放射延迟荧光。[18]如[14]~[17]中任一项所述的有机电致发光元件,其中,所述发光层含有所述π共轭类化合物和主体化合物。[19]如[14]~[17]中任一项所述的有机电致发光元件,其中,所述发光层含有所述π共轭类化合物和荧光发光性化合物及磷光发光性化合物的至少一个。[20]如[14]~[17]中任一项所述的有机电致发光元件,其中,所述发光层含有所述π共轭类化合物、荧光发光性化合物及磷光发光性化合物的至少一个和主体化合物。[21]如[18]或[20]所述的有机电致发光元件,其中,所述主体化合物具有下述通式(i)所表示的结构。[化学式4]通式(i)(通式(i)中,x101表示nr101、氧原子、硫原子、亚硫酰基、硫酰基、cr102r103或sir104r105,y1~y8分别独立地表示cr106或氮原子,r101~r106分别独立地表示氢原子或取代基,并且可以相互键合而形成环,ar101及ar102分别独立地表示任选被取代的芳基、或任选被取代的杂芳基,n101及n102各自表示0~4的整数。其中,r101为氢原子的情况下,n101表示1~4的整数。)[22]如[21]所述的有机电致发光元件,其中,具有所述通式(i)所表示的结构的主体化合物具有下述通式(ii)所表示的结构。[化学式5]通式(ii)(通式(ii)中,x101表示nr101、氧原子、硫原子、亚硫酰基、硫酰基、cr102r103或sir104r105,r101~r105分别表示氢原子或取代基,并且可以相互键合而形成环,ar101及ar102分别独立地表示任选被取代的芳基、或任选被取代的杂芳基,n102表示0~4的整数。)[23]一种显示装置,其含有[14]~[22]中任一项所述的有机电致发光元件。[24]一种照明装置,其含有[14]~[22]中任一项所述的有机电致发光元件。[25]一种有机电致发光元件,其含有阳极、阴极、及配置于所述阳极和所述阴极之间的有机层,所述有机层实质上由下述通式(201)所表示的π共轭类化合物构成。[化学式6](通式(201)(通式(201)中,a为吸电子基团,并且表示任选被取代的含氮芳香族6元环基,n1表示1~3的整数,n1为2个以上时,2个以上的所述a相互之间可以相同也可以不同,d为供电子基团,并且表示被供电子基团取代的芳基、任选被取代的氨基或烷基,n2表示2或3,2个以上的所述d相互之间可以相同也可以不同,且2个以上的所述d相互沿邻位配置)[26]如[25]所述的有机电致发光元件,其中,所述有机层为发光层。[27]如[26]所述的有机电致发光元件,其还含有配置于所述发光层和所述阳极之间的空穴传输层、及配置于所述发光层和所述阴极之间的电子传输层的至少一个。[28]如[26]或[27]所述的有机电致发光元件,其还含有配置于所述发光层和所述阳极之间的空穴注入层、以及配置于所述发光层和所述阴极之间的电子注入层的至少一个。[29]如[25]或[26]所述的有机电致发光元件,其中,所述有机层的一个面与所述阳极相接,并且所述有机层的另一个面与所述阴极相接。[30]一种有机电致发光元件,其含有阳极、阴极、以及一个面与所述阳极相接、且另一个面与所述阴极相接的有机层,所述有机层实质上由下述通式(201)所表示的π共轭类化合物和客体化合物构成。[化学式7](通式(201)(通式(201)中,a为吸电子基团,并且表示任选被取代的含氮芳香族6元环基,n1表示1~3的整数,n1为2个以上时,2个以上的所述a相互之间可以相同也可以不同,d为供电子基团,并且表示被供电子基团取代的芳基、任选被取代的氨基或烷基,n2表示2或3,2个以上的所述d相互之间可以相同也可以不同,且2个以上的所述d相互沿邻位配置)[31]如[30]所述的有机电致发光元件,其中,所述客体化合物为荧光发光性化合物(其中,不包括所述通式(201)所表示的π共轭类化合物)或磷光发光性化合物。[32]如[30]或[31]所述的有机电致发光元件,其中,所述客体化合物为荧光发光性化合物,所述荧光发光性化合物的含量为所述有机层的20体积%以下。[33]如[25]~[32]中任一项所述的有机电致发光元件,其中,所述通式(201)所表示的π共轭类化合物用下述通式(202)~(205)的任一种表示。[化学式8](通式(202)~(205)中,a1~a3分别与所述通式(201)中a的意义相同、d1~d3分别与所述通式(201)中d的意义相同)[34]如[25]~[33]中任一项所述的有机电致发光元件,其中,所述通式(201)中的d为下述通式(d-1)所表示的基团。[化学式9](通式(d-1)(通式(d-1)中,r1及r2分别表示任选被取代的芳基、或任选被取代的杂芳基,且r1及r2可以相互键合而形成环)[35]如[25]~[34]的任一种所述的有机电致发光元件,其中,所述通式(201)中的a为下述通式(a-1)或(a-2)所表示的基团。[化学式10](通式(a-1)或(a-2)中,x1~x3分别为-ch或氮原子,且至少一个为氮原子,z1~z3分别表示氢原子、氰基、任选被取代的芳基、或任选被取代的杂芳基)[36]如[25]~[35]中任一项所述的有机电致发光元件,其中,所述通式(201)所表示的π共轭类化合物用下述通式(206)或(207)表示。[化学式11](通式(206)或(207)中,z1及z2分别表示任选被取代的芳基)[37]如[25]~[35]中任一项所述的有机电致发光元件,其中,所述通式(201)所表示的π共轭类化合物用下述通式(208)表示。[化学式12](通式(208)中,d1~d3分别表示任选被取代的氨基)。[38]如[25]~[37]任一项所述的有机电致发光元件,其中,所述π共轭类化合物的最低激发单态能级和最低激发三重态能级的能量差的绝对值δest为0.5ev以下。[39]一种显示装置,其含有[25]~[38]的任一种所述的有机电致发光元件。[40]一种照明装置,其含有[25]~[38]的任一种所述的有机电致发光元件。发明效果通过本发明的上述技术方案,可以提供一种例如能够改良有机电致发光元件的发光效率的新型π共轭类化合物。另外,通过本发明的上述技术方案,可以提供一种实现了发光效率的改良的新型有机电致发光元件。另外,可以提供一种使用了该π共轭类化合物的有机电致发光元件材料、发光性薄膜、发光材料、以及具备该有机电致发光元件的显示装置及照明装置。另外,根据本发明,还可以提供一种有机el元件、以及具备该有机el元件的显示装置及照明装置,所述有机el元件不使发光效率和耐久性降低,可以使有机el元件的构成简单,生产率稳定性及可靠性高。附图说明图1是表示tadf化合物的能级图的示意图;图2是表示一般的荧光发光性化合物的能级图的示意图;图3是表示π共轭类化合物发挥辅助掺杂剂材料作用时的能级图的示意图;图4是表示π共轭类化合物发挥主体材料作用时的能级图的示意图;图5a是表示使用特定的π共轭类化合物的第1方式的有机el元件的构成例的图;图5b是表示使用特定的π共轭类化合物的第1方式的有机el元件的构成例的图;图5c是表示使用特定的π共轭类化合物的第1方式的有机el元件的构成例的图;图6是表示使用特定的π共轭类化合物的第2方式的有机el元件的构成例的图;图7是表示由有机el元件构成的显示装置的一例的示意图;图8是按照有源矩阵方式形成的显示装置的示意图;图9是表示像素电路的概略图;图10是按照无源矩阵方式形成的显示装置的示意图;图11是照明装置的概略图;图12是照明装置的示意图;图13是表示使用实施例中制作的本发明的π共轭类化合物得到的共蒸镀膜6-1中,在进行荧光减衰测定时从激发光照射起算的时间和光子数之间的关系的曲线图;图14是表示使用实施例中制作的本发明的π共轭类化合物得到的共蒸镀膜6-2中,在进行荧光减衰测定时从激发光照射起算的时间和光子数之间的关系的曲线图;图15是使用实施例中制作的本发明的π共轭类化合物而得到的共蒸镀膜6-3中,在进行荧光减衰测定时从激发光照射起算的时间和光子数之间的关系的曲线图;图16是使用实施例中制作的本发明的π共轭类化合物而得到的共蒸镀膜6-4中,在进行荧光减衰测定时从激发光照射起算的时间和光子数之间的关系的曲线图。标记说明10显示器13像素15扫描线16数据线17电源线20有机el元件21开关晶体管22驱动晶体管23电容器101照明装置内的有机el元件102玻璃盖板105阴极106有机el元件的构成层107带有透明电极的玻璃基板108氮气109捕水剂a显示部b控制部c配线部具体实施方式以下,对本发明和其构成要素、以及用于实施本发明的方式、方案进行详细的说明。需要说明的是,本申请中,“~”以含有其前后所记载的数值作为下限值及上限值的意思使用。本发明人等发现,在含有苯环、取代在其上的吸电子基团a和供电子基团d的π共轭类化合物中,通过将在苯环上取代的邻位组合中的至少一个设为“供电子基团d和供电子基团d”的组合、“吸电子基团a和吸电子基团a”的组合、“吸电子基团a和供电子基团d”的组合组合的任一种,可以提高例如有机电致发光元件的发光效率。其理由不一定清楚,但如下地推测。(同种之间的组合)取代在苯环上的供电子基团d(或供电子基团a)为2个以上时,与取代在苯环上的供电子基团d(或吸电子基团a)为1个相比,容易得到高的供电子性或高的吸电子性。另外,2个以上的供电子基团(或2个以上的吸电子基团a)取代在苯环的邻位;即,由于进行“空间上邻接”,因此,可以在激发状态下多个供电子基团(或吸电子基团)在空间上共振而将供电子性部所带的正电荷(或吸电子性部所带的负电荷)稳定化,因此,认为激发状态稳定化而无辐射失活得以抑制,可以提高发光效率。(不同种之间的组合)吸电子基团a和供电子基团d配置于苯环的邻位时,供电子基团d和吸电子基团a发生“空间上邻接”。由此,并不是从供电子基团d向吸电子基团a经由键合而形成ct性的激发状态,而是可以在位于极为相近的供电子基团d和吸电子基团a之间形成ct性的激发状态,ct性的激发状态稳定化,因此,认为发光效率升高。另外,在任一种方式中,认为吸电子基团a或供电子基团d不是通过贯穿键(throughbond)进行连结,而是通过贯穿空间来相互作用,因此,可以抑制吸收光谱或发光光谱的长波长化。(关于特定的π共轭类化合物)另外,如上所述,现有已知的荧光发光性化合物及磷光发光性化合物在以高浓度存在于有机el元件的发光层的情况下,促进无辐射失活而发光效率降低。因此,即使由荧光发光性化合物或磷光发光性化合物单独地构成发光层,也只能得到低的发光效率。其认为原因是,荧光发光性化合物及磷光发光性化合物以高浓度存在时,在激发状态下通过分子间相互作用而在多个分子间形成稳定结构,无辐射失活得以促进。因此,为了提高发光效率,更需要主体化合物。另外,作为现有已知的tadf化合物的4czipn或2czpn即使在单独包含在发光层中,也与上述的荧光发光性化合物或磷光发光性化合物的情况同样地,在高浓度下会促进无辐射失活,不能得到高的发光效率。与此相对,特定的π共轭类化合物(通式(201)所表示的π共轭类化合物)即使在发光层中以高浓度存在,也可具有高的发光效率。其理由如下地推测。即,在含有苯环、取代于其上的吸电子基团(通式(201)的a)和多个供电子基团(通式(201)的d)的特定π共轭类化合物中,将取代在苯环上的多个供电子基团(d)的取代位置设为邻位,由此,如上所述,采用在分子内它们在空间上接近的配置,可以在激发状态下多个供电子基团(d)在空间上共振而将供电子基团(d)所带的正电荷稳定化。其结果,不易引起分子间的相互作用,抑制无辐射失活,可以提高发光效率。这样,特定的π共轭类化合物即使在发光层中单独存在也具有高的发光效率,因此,目前认为需要的主体化合物变得不需要。另外,将取代在特定π共轭性化合物的苯环上的吸电子基团(a)设为“含氮芳香族6元环”,可以得到高电子传输性。这样,特定π共轭类化合物中,取代在苯环的邻位上的多个的供电子基团(d)负责分子间的空穴输送,取代在苯环上且由含氮芳香族6元环构成的吸电子基团(a)负责分子间的电子输送。由此,即使实质上仅由特定的π共轭类化合物构成配置于阳极和阴极之间的有机层,也可以在有机层中良好地输送空穴及电子,并进行再键合而发光。另外,现有已知的tadf化合物即dmac-dps即使单独包含在发光层中时也可以发光,但硫酰基容易分解,元件的耐久性低。与此相对,使用通式(201)所表示的特定π共轭类化合物的元件的耐久性也优异。通式(201)所表示的π共轭类化合物中所含的吸电子基团(a)即“含氮芳香族6元环基”具有难以分解的稳定的结构,且分子内的多个供电子基团(d)在激发状态下共振而形成稳定结构,因此,认为不易分解。这样,通过将通式(201)所表示的特定的π共轭类化合物用于发光层,可以使有机电致发光元件的构成变得简单,且可以提高耐久性。由此,可以提高有机el元件的生产稳定性及可靠性。另外,在通式(201)所表示的特定的π共轭类化合物中,多个供电子基团(d)接近而存在,因此,容易产生经由空间的相互作用,而非经由分子链的相互作用。由此,认为也可以抑制吸收光谱或发光光谱的长波长化。1.π共轭类化合物本发明的π共轭类化合物优选具有下述通式(1)所表示的结构。[化学式13]通式(1)的z1~z6分别表示氢原子、氘原子、供电子基团d或吸电子基团a。其中,z1~z6不相互键合而形成环。从得到高的发光效率的观点出发,优选z1~z6中的至少两个中的一个为供电子基团d,另一个为吸电子基团a。z1~z6中的邻位组合z1和z2、z2和z3、z3和z4、z4和z5、z5和z6及z6和z1的至少一个组合优选为供电子基团d和供电子基团d、吸电子基团a和吸电子基团a、或供电子基团d和吸电子基团a的组合组合的任一种。从增强供电子性或吸电子性的观点、或在供电子基团和吸电子基团之间容易生成ct性的激发状态的观点出发,z1~z6中邻位的2个以上、优选3个以上优选为供电子基团d或吸电子基团a。另外,更优选3个以上的供电子基团d连续地配置于邻位(相邻的位置),或3个以上的吸电子基团a连续地配置于邻位。3个以上的供电子基团d(或3个以上的吸电子基团a)连续地配置于邻位时,抑制被夹在2个供电子基团d(或2个吸电子基团a)之间的供电子基团d(或吸电子基团a)的旋转运动,激发状态被稳定化。另外,从供电子基团d和吸电子基团a的平衡变得良好、容易生成ct性的激发状态的观点出发,特别优选3个供电子基团d连续地配置于邻位、和/或3个吸电子基团a连续地配置于邻位。(供电子基团d)供电子基团d可以为“被供电子基团取代的芳基”、“任选被取代的供电子性杂环基”、“任选被取代的氨基”、或“烷基”。d所表示的“被供电子基团取代的芳基”中的芳基优选为从碳原子数6~24的芳香族烃环中衍生而成的基团。这种芳香族烃环的例子包括:苯环、茚环、萘环、薁环、芴环、菲环、蒽环、苊烯环、联苯撑环、并四苯环、芘环、戊搭烯环、醋蒽烯环、庚搭烯环、二苯并萘(triphenylene)环、as-苯并二茚环、环、s-苯并二茚环、七曜烯环、1,8-苯嵌萘(phenalene)环、荧蒽环、苝环、醋菲烯环、联苯环、联三苯环及联四苯环等。其中,优选为苯环、萘环、芴环、菲环、蒽环、联苯撑环、环、芘环、二苯并萘环、环、荧蒽环、苝环、联苯环、及联三苯环。该芳基所具有的供电子基团的例子包括:烷基、烷氧基、任选被取代的氨基、及任选被取代的供电子性杂环基等。其中,优选任选被取代的氨基、及任选被取代的供电子性杂环基。烷基可以为直链、支链或环状的任一种,例如可以为碳原子数1~20的直链或支链的烷基、或碳原子数5~20的环状烷基。在烷基的例子包括:甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、环己基、2-乙基己基、正庚基、正辛基、2-己基辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基、以及正二十基等;优选为甲基、乙基、异丙基、叔丁基、环己基、2-乙基己基及2-己基辛基。烷氧基可以为直链、支链或环状的任一种,例如可以为碳原子数1~20的直链或支链的烷氧基、或碳原子数6~20的环状的烷氧基。烷氧基的例子包括:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、新戊氧基、正己氧基、环己氧基、正庚氧基、正辛氧基、2-乙基己氧基、壬氧基、癸氧基、3,7-二甲基辛氧基、正十一烷氧基、正十二烷氧基、正十三烷氧基、正十四烷氧基、2-正己基-正辛氧基、正十五烷氧基、正十六烷氧基、正十七烷氧基、正十八烷氧基、正十九烷氧基、正二十烷氧基等,优选为甲氧基、乙氧基、异丙氧基、叔丁氧基、环己氧基、2-乙基己氧基、2-己基辛氧基。任选被取代的氨基中的取代基的例子包括:烷基、以及用烷基任选被取代的芳基。烷基及芳基与上述的烷基及(被供电子基团取代的芳基中的)芳基分别意义相同。任选被取代的供电子性杂环基的例子包括:与后述的供电子性的杂环基同样的基团。d所表示的“任选被取代的供电子性杂环基”中的“供电子性杂环基”优选为从碳原子数4~24的供电子性的杂环中衍生而成的基团。这种杂环的例子包括:吡咯环、吲哚环、咔唑环、吲哚并吲哚环、9,10-二氢吖啶环、5,10-二氢吩嗪环、5,10-二氢二苯并氮杂希林环、吩噁嗪环、吩噻嗪环、二苯并噻吩环、苯并呋喃基吲哚环、苯并噻吩并吲哚环、吲哚并咔唑环、苯并呋喃基咔唑环、苯并噻吩并咔唑环、苯并噻吩并苯并噻吩环、苯并咔唑环、二苯并咔唑环、氮杂咔唑环、及二氮杂咔唑环等。其中,优选为咔唑环、氮杂咔唑环、二氮杂咔唑环、吲哚并吲哚环、9,10-二氢吖啶环、5,10-二氢吩嗪环、吩噁嗪环、吩噻嗪环、二苯并噻吩环、及苯并呋喃基吲哚环。进一步优选列举任选被取代的咔唑环、任选被取代的氮杂咔唑环、任选被取代的二氮杂咔唑环、任选被取代的9,10-二氢吖啶环、任选被取代的吩噁嗪环、任选被取代的吩噻嗪环、任选被取代的5,10-二氢吩嗪环。供电子性杂环基可以为相同的或不同的2个以上的上述杂环经由单键键合而成的基团。该杂环基可具有取代基的例子包括:烷基、及任选被烷基取代的芳基。烷基及芳基分别与上述的烷基及芳基意义相同。d所表示的“任选被取代的氨基”中的取代基的例子包括:烷基、任选被烷基取代的芳基。烷基及芳基分别可以为与上述的烷基及芳基相同的基团。d所表示的“烷基”与上述的烷基意义相同。其中,作为供电子性高的供电子基团d的实例,优选为“被供电子基团取代的芳基”、“任选被取代的供电子性杂环基”、或“任选被取代的氨基”。供电子基团d具体而言优选为被供电子基团取代的苯基、任选被取代的咔唑基、任选被取代的氮杂咔唑基、任选被取代的二氮杂咔唑基、任选被取代的9,10-二氢吖啶基、任选被取代的吩噁嗪基、任选被取代的吩噻嗪基、任选被取代的5,10-二氢吩嗪基、任选被取代的二苯基氨基、或任选被取代的二烷基氨基。进一步优选为任选被取代的咔唑基、任选被取代的氮杂咔唑基、任选被取代的二氮杂咔唑基、任选被取代的9,10-二氢吖啶基、任选被取代的二苯基氨基。(吸电子基团a)吸电子基团a可以为“氟原子”、“被氟原子取代的烷基”、“任选被取代的羰基”、“任选被取代的硫酰基”、“任选被取代的氧化膦基”、“任选被取代的硼烷基”、“任选被取代的芳基(优选任选被吸电子基团取代的芳基)”、或“任选被取代的吸电子性杂环基”。a所表示的“被氟原子取代的烷基”中的烷基与上述烷基意义相同。被氟取代的烷基的例子包括:-cf3、-cf2h、-ch2f、-cf2cf3等。a所表示的“任选被取代的羰基”、“任选被取代的硫酰基”、“任选被取代的氧化膦基”及“任选被取代的硼烷基”中的取代基的例子包括:氘原子、氟原子、氰基、任选被氟原子取代的烷基、任选被取代的芳基、任选被取代的吸电子性杂环基。a所表示的“任选被取代的芳基”中的芳基与上述芳基意义相同。该芳基可具有的取代基的例子包括:氘原子、氟原子、氰基、任选被氟取代的烷基、任选被取代的羰基、任选被取代的硫酰基、任选被取代的氧化膦基、任选被取代的硼烷基、任选被取代的吸电子性的杂环基、任选被取代的氨基。其中,该芳基可具有的取代基优选为吸电子基团,优选为氟原子、氰基、任选被氟取代的烷基、任选被取代的羰基、任选被取代的硫酰基、任选被取代的氧化膦基、任选被取代的硼烷基、及任选被取代的吸电子性的杂环基;更优选为氟原子、氰基、任选被氟取代的烷基、及任选被取代的吸电子性的杂环基。a所表示的“任选被取代的吸电子性的杂环基”优选为从碳原子数3~24的吸电子性的杂环中衍生的基团。这种杂环基的例子包括:二苯并噻吩氧化物环、二苯并噻吩二氧化物环、吡啶环、哒嗪环、嘧啶环、吡嗪环、三嗪环、喹啉环、异喹啉环、喹唑啉环、1,2-二氮杂萘环、喹喔啉环、酞嗪环、蝶啶环、菲啶环、菲绕啉环、二苯并呋喃环、二苯并噻咯环、二苯并硼杂环戊二烯(dibenzoborole)环、二苯并磷杂环戊二烯氧化物(dibenzophospholeoxide)环等。其中,优选为吡啶环、嘧啶环、吡嗪环、三嗪环、喹啉环、异喹啉环、喹唑啉环、喹喔啉环、邻二氮杂菲环、菲绕啉环、二苯并呋喃环、二苯并硼杂环戊二烯环、二苯并磷杂环戊二烯氧化物环,特别优选为含氮芳香族6元环。需要说明的是,吸电子性杂环基可以是连结相同或不同的2个以上的上述杂环而成的基团。该吸电子性杂环基可具有的取代基的例子包括:氘原子、氟原子、氰基、任选被氟取代的烷基、任选被“任选被氟取代的烷基”取代的芳基、任选被氟取代的芳基,优选为氟原子、氰基、任选被氟取代的烷基、及任选被氟取代的芳基。作为吸电子性高的吸电子基团a的实例,特别优选被氰基取代的芳基、未取代的含氮芳香族6元环基、被氟原子取代的含氮芳香族6元环基、被氰基取代的含氮芳香族6元环基、被氟取代烷基取代的含氮芳香族6元环基、或被任选被“任选被取代的芳基”取代的含氮芳香族6元环基。含氮芳香族6元环的例子包括:吡啶环、哒嗪环、嘧啶环、吡嗪环、三嗪环、喹啉环、异喹啉环、喹唑啉环、邻二氮杂萘环、喹喔啉环、酞嗪环、蝶啶环、邻二氮杂菲环、菲绕啉环。其中,优选为吡啶环、嘧啶环、吡嗪环、三嗪环。含氮芳香族6元环也可以是连结相同或不同的2个以上的上述含氮芳香族6元环而成的环。作为更优选吸电子基团a,可举出被氰基取代的苯基、被氰基取代的吡啶基、任选被取代的噻嗪基、任选被取代的嘧啶基、任选被取代的三嗪基,进一步优选为对氰基苯基、间氰基苯基、邻氰基苯基、间二氰基苯基、2-氰基吡啶基、3-氰基吡啶基、2,6-二氰基吡啶基、2-氰基噻嗪基、2-氰基嘧啶基、5-氰基嘧啶基、任选被取代的2,4-二苯基嘧啶基、或任选被取代的二苯基三嗪基。π共轭类化合物优选用下述通式(2)~(10)、或(101)中的任一种表示。[化学式14]通式(2)~(10)或(101)的d1~d5分别独立地表示供电子基团d。供电子基团d与通式(1)的供电子基团d意义相同。通式(2)~(10)或(101)的z1~z5分别独立地表示氢原子、氘原子或吸电子基团a。吸电子基团a与通式(1)的吸电子基团a意义相同。在通式(2)~(10)或(101)中,优选满足d1=d2=d3=d4=d5和z1=z2=z3=z4=z5的至少一个。这是因为,不仅合成比较容易,而且容易得到良好的发光效率。π共轭类化合物更优选用下述通式(11)~(42)的任一种表示。[化学式15]通式(11)~(42)的d1~d5分别独立地表示供电子基团d。供电子基团d与通式(1)的供电子基团d意义相同。通式(11)~(42)的a1~a5分别独立地表示吸电子基团a。吸电子基团a与通式(1)的吸电子基团a意义相同。通式(11)~(42)中,优选满足d1=d2=d3=d4=d5和a1=a2=a3=a4=a5的至少一个。另外,通式(11)~(42)所表示的化合物中,从激发状态的稳定性的观点出发,优选3个以上的供电子基团d连续地配置于邻位、或3个以上的吸电子基团a连续地配置于邻位的化合物。具体而言,更优选为(11)~(15)、(18)、(19)、(23)、(24)、(26)、(27)、(29)、(30)、(39)、(40)、或(42)表示的化合物。另外,特别优选3个供电子基团d连续地配置于邻位、和/或3个吸电子基团a连续地配置于邻位,特别优选为通式(13)、(15)、(18)、(19)、(23)、(24)、(27)、(30)、(40)、或(42)所表示的化合物。具有所述通式(1)所表示的结构的π共轭类化合物可以作为含有该π共轭类化合物的有机电致发光元件材料、及含有该π共轭类化合物的发光性薄膜、及含有该π共轭类化合物的有机电致发光元件使用。具有所述通式(1)所表示的结构的π共轭类化合物可以通过电场激发而成为激发状态,生成激发子。因此,具有所述通式(1)所表示的结构的π共轭类化合物优选包含在有机电致发光元件的发光层中。即,从高发光性的观点出发,发光层优选单独使用具有所述通式(1)所表示的结构的π共轭类化合物;或含有具有所述通式(1)所表示的结构的π共轭类化合物和后述的主体化合物;或含有具有所述通式(1)所表示的结构的π共轭类化合物,并含有后述的荧光发光性化合物及磷光发光性化合物中的至少1种;或含有具有所述通式(1)所表示的结构的π共轭类化合物,并含有后述的荧光发光性化合物及磷光发光性化合物中的至少1种,以及还有后述的主体化合物。在具有所述通式(1)所表示的结构的π共轭类化合物中,该π共轭类化合物的最低激发单态能级和最低激发三重态能级的能量差的绝对值(δest)优选为0.50ev以下,优选包含在所述有机电致发光元件、所述元件材料、或所述发光性薄膜中。优选在照明装置及显示装置中具备本发明的有机电致发光元件。以下,举出本发明的π共轭类化合物的优选的具体例,但这些化合物有时进一步具有取代基,或存在结构异构体等,并不限定于本记载。[化学式16][化学式17][化学式18][化学式19][化学式20][化学式21][化学式22][化学式23][化学式24][化学式25][化学式26][化学式27][化学式28][化学式29][化学式30][化学式31][化学式32][化学式33][化学式34][化学式35][化学式36][化学式37][化学式38][化学式39][化学式40][化学式41][化学式42][化学式43][化学式44][化学式45][化学式46][化学式47][化学式48][化学式49][化学式50][化学式51][化学式52][化学式53][化学式54][化学式55][化学式56][化学式57][化学式58][化学式59][化学式60][化学式61][化学式62][化学式63][化学式64][化学式65][化学式66][化学式67][化学式68][化学式69][化学式70][化学式71][化学式72][化学式73][化学式74][化学式75][化学式76][化学式77]这些化合物中δest的绝对值为0.50ev以下的材料显示tadf性(延迟荧光性),即可以放射延迟荧光。在此,显示延迟荧光性是指:在进行荧光减衰测定时,所放射的荧光的减衰速度不同的成分为2种以上。需要说明的是,减衰慢的成分一般而言减衰时间多为亚微秒以上,减衰时间因材料不同而不同,因此,减衰时间没有限定。荧光减衰的测定一般而言可以如下地进行。对π共轭类化合物(发光性化合物)的溶液或薄膜、或π共轭类化合物和第二成分的共蒸镀膜在氮气氛下照射激发光,测量某发光波长的光子数。而且,此时,在所放射的荧光的减衰速度不同的成分为2种以上的情况下,π共轭类化合物为显示延迟荧光性的物质。需要说明的是,这些化合物具有双极性,可以对应于各种能级,因此,不仅可以作为发光主体使用,也可以作为适于空穴输送、电子输送的化合物使用,因此,并不限于在发光层中使用,也可以用于上述空穴注入层、空穴传输层、电子阻挡层、空穴阻挡层、电子传输层、电子注入层、中间层等。<特定的π共轭类化合物>本发明的π共轭类化合物中,作为即使在有机el元件的发光层中以高浓度存在也具有高发光效率的特定的π共轭类化合物,可举出下述通式(201)所表示的化合物。[化学式78]通式(201)的a为吸电子基团,并且表示任选被取代的含氮芳香族6元环基。“任选被取代的含氮芳香族6元环基”中的含氮芳香族6元环是碳原子数为3~13、优选3~5的吸电子性含氮芳香族6元环。含氮芳香族6元环可以为单环化合物,也可以为稠环化合物。含氮芳香族6元环为稠环化合物的情况下,其键是从构成稠环的环中含有氮原子的6元环中伸出的键。含氮芳香族6元环的实例包括:吡啶环、哒嗪环、嘧啶环、吡嗪环、三嗪环、喹啉环、异喹啉环、喹唑啉环、邻二氮杂萘环、喹喔啉环、酞嗪环、蝶啶环、邻二氮杂菲环、菲绕啉环。其中,优选吡啶环、嘧啶环、吡嗪环、三嗪环。含氮芳香族6元环可以是连结相同或不同的2个以上上述含氮芳香族6元环而形成的环。含氮芳香族6元环基可具有的取代基的例子包括:氟原子、氰基、任选被氟原子取代的烷基、任选被取代的芳基、以及任选被取代的杂芳基。含氮芳香族6元环基可具有的取代基的碳原子数的上限值例如可以为30,优选为15。“任选被氟原子取代的烷基”中的烷基可以为直链、支链或环状的任一种,例如可以为碳原子数1~20的直链或支链的烷基、或碳原子数5~20的环状烷基。烷基的例子包括:甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、环己基、2-乙基己基、正庚基、正辛基、2-己基辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基、及正二十基等;优选为甲基、乙基、异丙基、叔丁基、环己基、2-乙基己基及2-己基辛基。“任选被取代的芳基”中的芳基优选为从碳原子数6~24的芳香族烃环中衍生出来的基团。这种芳香族烃环的例子包括:苯环、茚环、萘环、薁环、芴环、菲环、蒽环、苊烯环、联苯撑环、并四苯环、芘环、戊搭烯环、醋蒽烯环、庚搭烯环、二苯并萘(triphenylene)环、as-苯并二茚环、环、s-苯并二茚环、七曜烯环、1,8-苯嵌萘(phenalene)环、荧蒽环、苝环、醋菲烯环、联苯环、联三苯环及联四苯环等。其中,优选为苯环、萘环、芴环、菲环、蒽环、联苯撑环、环、芘环、二苯并萘环、环、荧蒽环、苝环、联苯环、及联三苯环。芳基可具有的取代基的例子包括:任选被氟原子取代的烷基、烷氧基、氰基、杂芳基、芳氧基、氟原子、烷基磺酰基等。“任选被取代的杂芳基”中的杂芳基为碳原子数3~13、优选3~5的杂芳基,其例子包括:作为上述含氮芳香族6元环基列举的基团或三唑基等。杂芳基可具有的取代基的例子包括:任选被氟原子取代的烷基、烷氧基、氰基、芳基、芳氧基、芳基羰基等。任选被取代的含氮芳香族6元环基优选用通式(a-1)或(a-2)表示。[化学式79]通式(a-1)或(a-2)的x1~x3独立地为-ch或氮原子,至少一个表示氮原子。在得到良好的吸电子性的观点上,优选x1~x3的2个以上为氮原子。通式(a-1)或(a-2)的z1~z3为氢原子,或与通式(201)的含氮芳香族6元环基可具有的取代基意义相同。其中,z1~z3独立地优选为氢原子、氰基、任选被取代的芳基、或任选被取代的杂芳基,更优选为任选被取代的芳基。z1~z3相互之间可以相同或不同。通式(201)的n1表示1~3的整数。n1为2个以上时,2个以上的a相互相互之间可以相同也可以不同。通式(201)的d为供电子基团,并且表示“任选被取代的氨基”、“烷基”或“被供电子基团取代的芳基”。“任选被取代的氨基”中的取代基的例子包括:任选被取代的芳基、以及任选被取代的杂芳基。“任选被取代的氨基”中的取代基的碳原子数的上限值例如可以为30,优选为20。这些取代基可以相互键合而形成环,也可以不形成环,但从容易得到良好的供电子性的观点出发,优选形成环。环可以为5元环、6元环及7元环的任一种,优选为5元环或6元环。构成环的原子可以还含有氮原子以外的杂原子(例如氧原子、硫原子或硅原子)。“烷基”与作为上述含氮芳香族6元环基可具有的取代基而被列举的“任选被氟原子取代的烷基”中的烷基意义相同。“被供电子基团取代的芳基”中的芳基与作为上述含氮芳香族6元环基可具有的取代基而被列举的“任选被取代的芳基”中的芳基同样。该芳基可具有的供电子基团的例子包括:烷基、烷氧基、以及任选被取代的氨基,优选为任选被取代的氨基。作为供电子基团的烷基及任选被取代的氨基与上述的烷基及任选被取代的氨基分别意义相同。作为供电子基团的烷氧基可以为直链、支链或环状的任一种,例如可以为碳原子数1~20的直链或支链的烷氧基、或碳原子数6~20的环状烷氧基。烷氧基的例子包括:甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、新戊氧基、正己氧基、环己氧基、正庚氧基、正辛氧基、2-乙基己氧基、壬氧基、癸氧基、3,7-二甲基辛氧基、正十一烷氧基、正十二烷氧基、正十三烷氧基、正十四烷氧基、2-正己基-正辛氧基、正十五烷氧基、正十六烷氧基、正十七烷氧基、正十八烷氧基、正十九烷氧基、正二十烷氧基等,优选为甲氧基、乙氧基、异丙氧基、叔丁氧基、环己氧基、2-乙基己氧基、2-己基辛氧基。其中,从供电子性高方面考虑,通式(201)中的d优选为任选被取代的氨基,优选为通式(d-1)所表示的基团。[化学式80]通式(d-1)的r1及r2分别表示任选被取代的芳基、或任选被取代的杂芳基。任选被取代的芳基与上述“被供电子基团取代的芳基”意义相同任选被取代的杂芳基中的“杂芳基”为碳原子数3~8的杂芳基,其例子包括:苯并噻吩基、苯并呋喃基、吡啶基等。杂芳基可具有的取代基的例子包括:烷基、任选被取代的氨基等。r1及r2可以相互键合而形成环,也可以不形成环,但从容易得到良好的供电子性的观点出发,优选形成环。r1及r2相互键合而形成的环与在上述的d所表示的“任选被取代的氨基”中取代基相互键合而形成的环意义相同。通式(d-1)所表示的基团的例包括:从二苯基氨基、二吡啶氨基、吲哚环、咔唑环、吲哚并吲哚环、9,10-二氢吖啶环、吩噁嗪环、吩噻嗪环、吲哚并咔唑环、苯并呋喃基咔唑环、苯并噻吩并咔唑环、苯并咔唑环、二苯并咔唑环、氮杂咔唑环、及二氮杂咔唑环、苯并呋喃基吲哚环、苯并噻吩并吲哚环中衍生出来的基团。通式(201)的n2表示2或3。其中,至少2个d相互沿邻位配置。由此,取代在苯环上的多个供电子基团d在空间上接近,因此,可以在激发状态下多个供电子基团空间上发生共振而对供电子性部所带的正电荷稳定化。至少2个d相互之间可以相同也可以不同。通式(201)所表示的π共轭类化合物优选用下述通式(202)~(205)的任一种表示。[化学式81]通式(202)~(205)的a1~a3分别与通式(201)的a意义相同,通式(202)~(205)的d1~d3分别与通式(201)的d意义相同。a1~a3相互之间可以相同也可以不同。通式(202)、(203)及(205)中,从在激发状态下容易实现共振稳定化的观点出发,优选a1~a3中的2个以上为相同。d1~d3相互之间可以相同也可以不同。从在激发状态下容易实现共振稳定化的观点出发,优选d1~d3中的2个以上为相同。其中,从使homo和lumo分离集中存在而容易得到tadf性的观点出发,优选通式(204)所表示的化合物。通式(201)所表示的π共轭类化合物更优选用下述通式(206)~(208)的任一种表示。[化学式82]通式(206)及(207)的z1及z2分别与通式(a-1)或(a-2)的z1~z3分别意义相同,优选为任选被取代的芳基。在z1及z2的组合的例子包括如下。[化学式83][化学式84][化学式85][化学式86][化学式87][化学式88]通式(208)的d1~d3优选为任选被取代的氨基。任选被取代的氨基与通式(201)的d所表示的“任选被取代的氨基”意义相同。d1~d3的组合的例子包括如下。[化学式89][化学式90][化学式91][化学式92][化学式93]通式(201)所表示的π共轭类化合物的例子包括如下。[化学式94][化学式95]通式(201)所表示的π共轭类化合物的最低激发单态能级和最低激发三重态能级的能量差的绝对值(δest)优选为0.50ev以下。这种π共轭类化合物可显示良好的tadf性。通式(201)所表示的π共轭类化合物优选包含在有机电致发光元件、所述元件材料、或所述发光性薄膜中。通式(201)所表示的π共轭类化合物包含在有机电致发光元件的发光层中。即,从高发光性的观点出发,发光层优选实质上由通式(201)所表示的π共轭类化合物构成,或实质上由通式(201)所表示的π共轭类化合物和客体化合物构成。优选在照明装置及显示装置中具有使用通式(201)所表示的π共轭类化合物的有机电致发光元件。另外,通式(201)所表示的π共轭类化合物具有双极性,可以对应于各种能级,因此,不仅可以作为主体化合物使用,也可以作为适于空穴输送、电子输送的化合物使用,因此,并不限于发光层中的使用,也可以用于上述的空穴注入层、空穴传输层、电子阻挡层、空穴阻挡层、电子传输层、电子注入层、中间层等。<合成方法>上述通式(1)所表示的π共轭类化合物(也包含通式(201)所表示的化合物)例如可以通过参照国际公开第2010/113755号、organicletters,2002,4,1783-1785.、angew.chem.int.ed.2010,49,2014-2017.中记载的方法、或这些文献中记载的参考文献中记载的方法来合成。下面,对与本发明的技术思想关联的有机el的发光方式及发光材料进行说明。<有机el的发光方式>作为有机el的发光方式,包括:从三重态激发状态返回到基态时发出光的“磷光发光”和从单态激发状态返回到基态时发出光的“荧光发光”这两种途径。在有机el那样的电场中激发的情况下,三重态激发子以75%的概率生成,单态激发子以25%的概率生成,因此,磷光发光与荧光发光相比,可以提高发光效率,这是对于实现低消费电力化而言是优异的方式。另一方面,在荧光发光中,以75%的概率生成,通常,激发子的能量由于无辐射失活而使那些只能变为热量的三重态激发子以高密度存在,由此,发现利用了由两个三重态激发子产生一个单态激发子而提高发光效率的tta(triplet-tripletannihilation,也称为,triplet-tripletfusion:简称为“ttf”。)机制的方式。另外,近年来,根据安达等的发现,通过缩小单态激发状态和三重态激发状态的能量间隙,根据发光中的焦耳热和/或发光元件所处的环境温度从能级低的三重态激发状态向单态激发状态引起逆系间穿越,结果,发现了可以进行大致接近于100%的荧光发光的现象(热激发型延迟荧光,或也称为热激发型延迟荧光:“tadf”)和可以产生该现象的荧光物质(例如参照非专利文献1等)。<磷光发光性化合物>如上所述,磷光发光在发光效率上与荧光发光相比,理论上为3倍,是有利的,但从三重态激发状态向单态基态的能量失活(=磷光发光)为禁戒跃迁,另外,同样地从单态激发状态向三重态激发状态的系间穿越也为禁戒跃迁,因此,通常其速度常数小。即,不易引起迁移,因此,激发子寿命从毫秒变长至秒级,难以得到所期望的发光。其中,在使用了铱或铂等重金属的络合物发光的情况下,由于中心金属的重原子效应,所述禁戒跃迁的速度常数增大3个数量级以上,在选择某些配体时,也可以得到100%的磷光量子收率。但是,为了得到这种理想的发光,需要使用作为稀有金属的铱或钯、铂等被称为所谓的铂属的贵金属,大量地被使用时,其埋藏量或金属自身的价格成为产业上大的问题。<荧光发光性化合物>一般的荧光发光性化合物没有必要采用像磷光发光性化合物这样的重金属络合物,可以使用由碳、氧、氮及氢等一般元素的组合构成的所谓有机化合物,另外,也可以使用磷、硫、硅等其它非金属元素,另外,还可以有效利用铝或锌等典型金属的络合物等,可以说其多样性几乎无限。其中,现有的荧光化合物如上所述只能适用于激发子的25%发光,因此,不期望磷光发光那样的高效率发光。<延迟荧光化合物>[激发三重态-三重态湮灭(tta)延迟荧光化合物]为了解决荧光发光性化合物的问题点而出现的是:利用了延迟荧光的发光方式。以三重态激发子彼此的冲突为起源的tta方式可以在如下所述的通式中记述。即,具有以下优点:以往激发子的能量由于无辐射失活而仅变换为热量的三重态激发子的一部分可以逆系间穿越为有助于发光的单态激发子,在实际的有机el元件中,可以得到现有的荧光发光元件的约2倍的外部导出量子效率。通式:t*+t*→s*+s(式中,t*表示三重态激发子,s*表示单态激发子,s表示基态分子。)但是,由上式也可以得知,可以由两个三重态激发子只能生成一个可以用于发光的单态激发子,因此,原理上不能以该方式得到100%的内部量子效率。[热活性型延迟荧光(tadf)化合物]作为另一个高效率荧光发光的tadf方式为可以解决tta的问题点的方式。荧光发光性化合物具有如上所述可以无限地进行分子设计的优点。即,在进行了分子设计的化合物中,特异性地存在三重态激发状态和单态激发状态的能级差极为接近的化合物。这种化合物虽然在分子内不具有重原子,但是,由于δest小,因此会引发通常不引发的从三重态激发状态单态向激发状态的逆系间穿越。另外,由于从单态激发状态向基态的失活(=荧光发光)的速度常数非常大,因此,三重态激发子与其自身在基态下热失活(无辐射失活)相比,一边经由单态激发状态发出荧光,一边返回到基态这一点在速度论上是有利的。因此,tadf理想而言可以进行100%的荧光发光。<与δest有关的分子设计思想>对用于缩小上述δest的分子设计进行说明。为了缩小δest,原理上缩小分子内的最高被占分子轨道(highestoccupiedmolecularorbital:homo)和最低空分子轨道(lowestunoccupiedmolecularorbital:lumo)的空间重叠是最有效的。一般而言,在分子的电子轨道中,已知有homo分布于给电子性部位,lumo分布于电子吸引性部位,通过在分子内导入给电子性和电子吸引性的骨架,可以疏远homo和lumo存在的位置。例如,在《迎接实用化阶段的有机光电子学》应用物理第82卷、第6号、2013年中,通过导入氰基、三嗪等吸电子性骨架和咔唑或二苯基氨基等给电子性骨架,使lumo和homo分别定域。另外,缩小化合物的基态和三重态激发状态的分子结构变化也是有效的。作为用于缩小结构变化的方法,例如使化合物变为刚直性等是有效的。在此进行叙述的刚直是指:例如抑制分子内的环与环的键的自由旋转,另外导入π共轭面大的稠环等在分子内自由地运动的部位少。特别是通过使参与发光的部位变得刚直,可以缩小激发状态中的结构变化。<tadf化合物存在的一般问题>tadf化合物从其发光机制及分子结构方面考虑,存在着各种问题。在tadf化合物中,为了缩小δest,需要使homo和lumo的存在的部位尽可能原理,因此,分子的电子状态成为接近于homo部位和lumo部位分离的供体/受体型的分子内ct(分子内电荷移动状态)的状态。这种分子存在多个时,使一个分子的供体部分和另一个分子的受体部分接近时,可谋求稳定化。这种稳定化状态不限于在2分子间形成,即使在3分子间或5分子间等多个分子间也可以形成,结果,通过具有宽广的分布的各种稳定化状态存在,吸收光谱及发光光谱的形状变得宽阔。另外,即使在没有形成超过2分子的多分子集合体的情况下,也因两个分子的相互作用的方向或角度等不同而取得各种存在状态,因此,基本上仍然吸收光谱及发光光谱的形状变得宽阔。发光光谱变得宽阔产生两个大的问题。一个问题是发光色的色纯度变低的问题。在使用于照明用途的情况下,不那么成为大的问题,但在用于电子显示器用途的情况下,色再现域变小,另外,纯色的色再现性变低,因此,实际上作为商品使用会变得困难。另一个问题在于,发光光谱的短波长侧的上升波长(称为“荧光0-0带”。)发生短波长化,即进行高s1化(最低激发单态能级的高能量化)。当然,荧光0-0带发生短波长化时,与s1相比,源自能量低的t1的磷光0-0带也发生短波长化(t1能级的提高)。因此,用于主体化合物的化合物不引起来自掺杂剂的逆能量移动,因此,产生高s1能级且高t1能级的需要。这是非常大的问题。基本上由有机化合物构成的主体化合物在有机el元件中采用阳离子自由基状态、阴离子自由基状态及激发状态这样的多种活性且不稳定的化学种的状态,但这些化学种可以通过扩大分子内的π共轭系而比较稳定地存在。但是,为了对分子赋予高的s1能级且高的t1能级,需要缩小或切断分子内的π共轭类,与稳定性并存变得困难,结果,会缩短发光元件的寿命。在不含有重金属的tadf化合物中,从三重态激发状态向基态失活的跃迁为禁戒跃迁,因此,三重态激发状态下的存在时间(激发子寿命)从数百μ秒向毫秒级非常长。因此,即使主体化合物的t1能级为比荧光发光性化合物的能量高的能级,由其存在时间的长度从荧光发光性化合物的三重态激发状态向主体化合物引起逆能量移动的概率会增大。其结果,不充分地引起本来意图的从tadf化合物的三重态激发状态向单态激发状态的逆系间穿越,而是不优选的向主体化合物的逆能量移动成为主流,产生不能得到充分的发光效率的不良情况。为了解决如上所述的问题,需要将tadf化合物的发光光谱形状尖锐化,缩小发光最大波长和发光光谱的上升波长之差。因此,基本上可以通过缩小单态激发状态及三重态激发状态的分子结构的变化而实现。另外,为了抑制向主体化合物的逆能量移动,缩短tadf化合物的三重态激发状态的存在时间(激发子寿命)是有效的。为了实现该目标,通过采取缩小基态和三重态激发状态的分子结构变化及为了解开禁戒跃迁而导入适合的取代基或元素等对策,可以解决问题点。在本发明的π共轭类化合物(通式(1)所表示的化合物)中,具有苯环、取代在其上的吸电子基团a和供电子基团d,将取代在苯环的邻位上的组合的至少一个设为“供电子基团d和供电子基团d”、“吸电子基团a和吸电子基团a”、“吸电子基团a和供电子基团d”的任一种。2个以上的供电子基团(或2个以上的吸电子基团a)在苯环的邻位上进行取代;即,进行“在空间上邻接”,因此,可以在激发状态下多个供电子基团(或吸电子基团)在空间上进行共振而将供电子性部所带的正电荷(或吸电子性部所带的负电荷)稳定化,因此,认为激发状态稳定化而抑制无辐射失活,可以提高发光效率。通过将吸电子基团a和供电子基团d配置于苯环的邻位,供电子基团d和吸电子基团a实现“空间上邻接”。由此,在位置非常接近的供电子基团d和吸电子基团a之间可以形成ct性的激发状态,并不是从供电子基团d向吸电子基团a经由键合形成ct性的激发状态,ct性的激发状态稳定化,因此,认为发光效率升高。另外,在任一种方式中,吸电子基团a或供电子基团d并非通过三键进行连结,而是通过贯穿空间来相互作用,因此,认为也可以抑制吸收光谱或发光光谱的长波长化。另外,特别是在含有苯环、取代在其上的吸电子基团(a)和多个供电子基团(d)的上述特定π共轭类化合物(通式(201)所表示的化合物)中,通过将取代在苯环上的多个供电子基团(d)的取代位置设为邻位,不易引起分子间的相互作用,抑制无辐射失活,可以提高发光效率。这样,特定的π共轭类化合物即使在发光层中单独存在,也具有高的发光效率,因此,目前需要的主体化合物变得不需要。另外,在该特定的π共轭类化合物中,通过将取代在苯环上的吸电子基团(a)设为“含氮芳香族6元环”,可以得到高的电子输送性。另外,在通式(201)所表示的特定π共轭类化合物中,由于多个供电子基团(d)接近而存在,因此,并非经由分子链的相互作用,容易产生经由空间的相互作用。由此,认为也可以抑制吸收光谱或发光光谱的长波长化。以下,对与本发明的π共轭类化合物有关的各种测定方法进行记载。[电子密度分布]从缩小δest的观点出发,本发明的π共轭系化合物优选在分子内homo和lumo实质上分离。关于这些homo及lumo的分布状态,可以由通过分子轨道计算而得到的结构最适化时的电子密度分布求出。在本发明中的π共轭系化合物的分子轨道计算的结构最适化及电子密度分布的计算中,作为计算方法,可以采用使用了作为泛函数的b3lyp、作为基函数的6-31g(d)的分子轨道计算用软件算出,对软件没有特别限定,使用任一种都可以同样地求出。在本发明中,作为分子轨道计算用软件,使用美国gaussian公司制成的gaussian09(revisionc.01,m.j.frisch,etal,gaussian,inc.,2010.)。另外,“homo和lumo实质上发生分离”是指:通过上述分子计算所算出的homo轨道分布及lumo轨道分布的中心部位分离,更优选homo轨道的分布和lumo轨道的分布几乎不重叠。另外,关于homo和lumo的分离状态,也可以由使用b3lyp作为上述的泛函数、使用6-31g(d)作为基底函数的结构最适化计算,进一步实施利用时间依赖密度泛函数法(time-dependentdft)的激发状态计算求出s1、t1的能级(分别e(s1)、e(t1)),算出δest=|e(s1)-e(t1)|。表示所算出的δest越小,homo和lumo越进一步分离。在本发明中,使用与上述同样的计算方法所算出的δest优选为0.50ev以下,更优选为0.30ev以下,进一步优选为0.10ev以下。[最低激发单态能级s1]关于本发明中的π共轭系化合物的最低激发单态能级s1,在本发明中,也用与通常的方法同样地算出的能级定义。即,将成为测定对象的化合物蒸镀在石英基板上来制作试样,在常温(300k)下测定该试样的吸收光谱(纵轴:吸光度、横轴:设为波长)。对该吸收光谱的长波长侧的上升点引切线,基于该切线和横轴的交点的波长值,由规定的换算式算出。其中,在本发明中使用的π共轭系化合物的分子自身的凝聚性比较高的情况下,有可能在薄膜的测定中因凝集导致误差。考虑到本发明中的π共轭系化合物的斯托克斯位移比较小、另外激发状态和基态的结构变化小,本发明中的最低激发单态能级s1使用室温(25℃)下的π共轭系化合物的溶液状态的最大发光波长的峰值作为近似值。在此,使用的溶剂可以使用对π共轭系化合物的凝聚状态不产生影响、即溶剂效果的影响小的溶剂、例如环己烷或甲苯等非极性溶剂等。[最低激发三重态能级t1]关于本发明中的π共轭系化合物的最低激发三重态能级(t1),根据溶液或薄膜的光致发光(pl)特性算出。例如,作为薄膜中的算出方法,可以在将稀薄状态的π共轭系化合物的分散物制成薄膜之后,使用超高速扫描照相机测定过渡pl特性,由此进行荧光成分和磷光成分的分离,将其能量差作为δest,由最低激发单态能级求出最低激发三重态能级。在测定、评价中,关于绝对pl量子收率的测定,使用绝对pl量子收率测定装置c9920-02(浜松光电社制造)。发光寿命使用超高速扫描照相机c4334(浜松光电社制造),一边使样品用激光激发,一边进行测定。《有机el元件的构成层》本发明的有机el元件是在阳极和阴极之间至少具有发光层的有机电致发光元件,其特征在于,至少1层该发光层含有具有所述通式(1)所表示的结构的π共轭类化合物。作为本发明的有机el元件中的代表的元件结构,可举出以下结构,但并不限定于这些。(1)阳极/发光层/阴极(2)阳极/发光层/电子传输层/阴极(3)阳极/空穴传输层/发光层/阴极(4)阳极/空穴传输层/发光层/电子传输层/阴极(5)阳极/空穴传输层/发光层/电子传输层/电子注入层/阴极(6)阳极/空穴注入层/空穴传输层/发光层/电子传输层/阴极(7)阳极/空穴注入层/空穴传输层/(电子阻挡层/)发光层/(空穴阻挡层/)电子传输层/电子注入层/阴极在上述中优选使用(7)的结构,但并不限定于此。用于本发明的发光层由单层或多层构成,发光层为多个的情况下,可以在各发光层之间设置非发光性的中间层。根据需要,可以在发光层和阴极之间设置空穴阻挡层(也称为空穴屏障层)或电子注入层(也称为阴极缓冲层),另外,也可以在发光层和阳极之间设置电子阻挡层(也称为电子屏障层)或空穴注入层(也称为阳极缓冲层)。本发明所使用的电子传输层为具有传输电子的功能的层,广义上,电子注入层、空穴阻挡层也包含在电子传输层中。另外,可以由多层构成。本发明所使用的空穴传输层为具有传输空穴的功能的层,广义上,空穴注入层、电子阻挡层也包含在空穴传输层中。另外,可以由多层构成。(串联结构)另外,本发明的有机el元件可以为叠层多个含有至少1层发光层的发光单元而成的、所谓串联结构元件。作为串联结构的代表性元件构成,可以列举例如以下的构成。阳极/第1发光单元/中间层/第2发光单元/中间层/第3发光单元/阴极在此,上述第1发光单元、第2发光单元及第3发光单元可以全部相同,也可以不同。另外,也可以两个发光单元相同,剩余的一个不同。多个发光单元可以直接叠层,也可以经由中间层而叠层,中间层一般而言也称为中间电极、中间导电层、电荷产生层、夺电子层、连接层、中间绝缘层,只要是具有对阳极侧的邻接层供给电子、对阴极侧的邻接层供给空穴的功能的层即可,可以使用公知的材料构成。作为中间层所使用的材料,可列举例如:ito(铟锡氧化物)、izo(铟锌氧化物)、zno2、tin、zrn、hfn、tiox、vox、cui、inn、gan、cualo2、cugao2、srcu2o2、lab6、ruo2、al等导电性无机化合物层、或au/bi2o3等2层膜、或sno2/ag/sno2、zno/ag/zno、bi2o3/au/bi2o3、tio2/tin/tio2、tio2/zrn/tio2等多层膜、另外c60等富勒烯类、低聚噻吩等导电性有机物层、金属酞菁类、无金属酞菁类、金属卟啉类、无金属卟啉类等导电性有机化合物层等,但本发明并不限定于这些。作为发光单元内的优选结构,可列举例如从上述的代表性元件构成中列举的(1)~(7)的结构中除去了阳极和阴极的结构等,但本发明并不限定于这些。作为串联型有机el元件的具体例,可列举例如:美国专利第6337492号说明书、美国专利第7420203号说明书、美国专利第7473923号说明书、美国专利第6872472号说明书、美国专利第6107734号说明书、美国专利第6337492号说明书、国际公开第2005/009087号、日本特开2006-228712号公报、日本特开2006-24791号公报、日本特开2006-49393号公报、日本特开2006-49394号公报、日本特开2006-49396号公报、日本特开2011-96679号公报、日本特开2005-340187号公报、专利第4711424号公报、专利第3496681号公报、专利第3884564号公报、专利第4213169号公报、日本特开2010-192719号公报、日本特开2009-076929号公报、日本特开2008-078414号公报、日本特开2007-059848号公报、日本特开2003-272860号公报、日本特开2003-045676号公报、国际公开第2005/094130号等中记载的元件构成或构成材料等,但本发明并不限定于这些。以下,对构成本发明的有机el元件的各层进行说明。《发光层》本发明中所使用的发光层是提供从电极或邻接层中注入的电子及空穴再键合并经由激发子而发光的场所的层,发光的部分可以为发光层的层内,也可以为发光层和邻接层的界面。本发明所使用的发光层只要满足本发明中规定的主要条件,对其构成没有特别限制。发光层的层厚的总和没有特别限制,从形成的膜的均质性、或防止施加发光时并不需要的高电压、且提高对于驱动电流的发光色的稳定性的观点出发,优选调整为2nm~5μm的范围,更优选调整为2~500nm的范围,进一步优选调整为5~200nm的范围。另外,作为本发明所使用的每个发光层的层厚,优选调整为2nm~1μm的范围,更优选调整为2~200nm的范围,进一步优选调整为3~150nm的范围。用于本发明的发光层可以为一层,也可以由多层构成。将本发明的π共轭类化合物用于发光层的情况下,可以单独使用,也可以与后述的主体材料、荧光发光材料、磷光发光材料等混合而使用。例如,可以将发光层的至少一层设为含有发光掺杂剂(也称为发光性化合物、发光性掺杂剂,也简称为掺杂剂)、还含有主体化合物(也称为基质材料、发光主体化合物,也简称为主体)的层。至少一层发光层含有本发明的π共轭类化合物和主体化合物时,发光效率提高,因此优选。另外,至少一层发光层含有本发明的π共轭类化合物并含有荧光发光性化合物及磷光发光性化合物中的至少1种时,发光效率提高,因此优选。至少一层发光层含有本发明的π共轭类化合物,并含有荧光发光性化合物及磷光发光性化合物中的至少1种,还含有主体化合物时,发光效率提高,因此优选。(1)发光掺杂剂作为发光掺杂剂,优选使用荧光发光性掺杂剂(也称为荧光发光性化合物、荧光掺杂剂)和磷光发光性掺杂剂(也称为磷光发光性化合物、磷光掺杂剂)。在本发明中,发光层优选在0.1~50质量%的范围内含有本发明的π共轭类化合物作为荧光发光性化合物或辅助掺杂剂,上述含量特别优选在1~30质量%的范围内。在本发明中,发光层优选在0.1~50质量%的范围内,特别优选在1~30质量%的范围内含有发光性化合物。关于发光层中的发光性化合物的浓度,可以基于所使用的特定发光性化合物及装置的必要条件任意地确定,在发光层的层厚方向上,可以以均匀的浓度存在,另外也可以具有任意的浓度分布。另外,本发明中所使用的发光性化合物可以组合使用多种,也可以将结构不同的荧光发光性化合物彼此的组合、或荧光发光性化合物和磷光发光性化合物组合而使用。由此,可以得到任意的发光色。在发光层中含有最低激发单态能级和最低激发三重态能级的差的绝对值(δest)为0.50ev以下的本发明的π共轭类化合物、发光性化合物和主体化合物时,本发明的π共轭类化合物作为辅助掺杂剂起作用。另一方面,发光层含有本发明的π共轭类化合物和发光性化合物、不含有主体化合物时,本发明的π共轭类化合物作为主体化合物起作用。发光层仅含有本发明的π共轭类化合物时,本发明的π共轭类化合物作为主体化合物和发光性化合物起作用。作为效果显现的机理,任何情况都是同样的,其机理在于将本发明的π共轭类化合物上生成的三重态激发子通过逆系间穿越(risc)向单态激发子变换这一点上。由此,可以将在本发明的π共轭类化合物上生成的理论上全部的激发子能量进行荧光共振能量移动(fret)至发光性化合物,可以表现出高发光效率。因此,发光层含有本发明的π共轭类化合物、发光性化合物及主体化合物这3种成分时,优选π共轭类化合物的s1和t1的能级比主体化合物的s1和t1的能级低,比发光性化合物的s1和t1的能级高。同样地,发光层含有本发明的π共轭类化合物和发光性化合物这2种成分时,优选π共轭类化合物的s1和t1的能级比发光性化合物的s1和t1的能级高。图3及图4示出本发明的π共轭类化合物分别发挥辅助掺杂剂及主体化合物的作用时的示意图。图3及图4为一例,在本发明的π共轭类化合物上生成的三重态激发子的生成过程并不仅限定于电场激发,也包含来自发光层内或周边层界面的能量移动或电子移动等。另外,图3及图4中,使用荧光发光性化合物作为发光材料,但并不限定于此,可以使用磷光发光性化合物,也可以使用荧光发光性化合物和磷光发光性化合物这两者。将本发明的π共轭类化合物用作辅助掺杂剂时,优选发光层相对于π共轭类化合物以质量比计含有100%以上的主体化合物,荧光发光性化合物和/或磷光发光性化合物相对于π共轭类化合物的含量以质量比为0.1~50%的范围内。将本发明的π共轭类化合物用作主体化合物时,优选发光层相对于π共轭类化合物在质量比0.1~50%的范围内含有荧光发光性化合物和/或磷光发光性化合物。将本发明的π共轭类化合物用作辅助掺杂剂或主体化合物时,优选本发明的π共轭类化合物的发光光谱和发光性化合物的吸收光谱重叠。本发明的有机el元件或本发明所使用的化合物的发光颜色如下确定:在“新编色彩科学手册”(日本色彩学会编、东京大学出版会、1985)的108页的图3.16中,将利用分光辐射亮度计cs-1000(柯尼卡美能达(株)制造)测定的结果对照cie色度坐标,通过对照时确定的颜色来确定。在本发明中,还优选1层或多层的发光层含有发光色不同的多种发光掺杂剂,显示白色发光。关于显示白色的发光掺杂剂的组合,没有特别限定,可举出例如蓝和橙、或蓝、绿和红的组合等。所谓本发明的有机el元件中的白色,优选的是,在通过上述方法测定2度视场角正面亮度时,在1000cd/m2的cie1931表色系中的色度位于x=0.39±0.09、y=0.38±0.08的区域内。(1.1)荧光发光性掺杂剂荧光发光性掺杂剂(荧光掺杂剂)可以使用本发明的π共轭类化合物,可以从有机el元件的发光层中所使用的公知的荧光掺杂剂或延迟荧光性掺杂剂中适当选择来使用。作为可以在本发明中使用的公知的荧光掺杂剂的具体例子,可举出例如:蒽衍生物、芘衍生物、衍生物、荧蒽衍生物、苝衍生物、芴衍生物、芳基乙炔衍生物、苯乙烯基亚芳基衍生物、苯乙烯基胺衍生物、芳基胺衍生物、硼络合物、香豆素衍生物、吡喃衍生物、花青苷衍生物、克酮酸衍生物、方酸内鎓盐衍生物、氧代苯并蒽衍生物、荧光素衍生物、若丹明衍生物、吡喃鎓衍生物、苝衍生物、聚噻吩衍生物、或稀土络合物系化合物等。另外,近年来,也开发了采用延迟荧光的发光掺杂剂,可以使用这些物质。作为采用延迟荧光的发光掺杂剂的具体例子,可举出例如:国际公开第2011/156793号、日本特开2011-213643号公报、日本特开2010-93181号公报、日本专利5366106号公报等中记载的化合物,但本发明并不限定于这些。(1.2)磷光发光性掺杂剂对用于本发明的磷光发光性掺杂剂进行说明。用于本发明的磷光发光性掺杂剂被定义为观测到来自激发三重态的发光的化合物,具体而言,是在室温(25℃)下进行磷光发光的化合物,磷光量子收率在25℃下为0.01以上的化合物,但优选的磷光量子收率为0.1以上。上述磷光量子收率可以通过第4版实验化学讲座7的分光ii的398页(1992年版,丸善)中记载的方法进行测定。溶液中的磷光量子收率可以使用各种溶剂进行测定,但用于本发明的磷光掺杂剂只要在任意的溶剂的任一种中实现上述磷光量子收率(0.01以上)即可。磷光掺杂剂可以从有机el元件的发光层中所使用的公知的物质中适当选择而使用。作为可以在本发明中使用的公知的磷光掺杂剂的具体例子,可举出以下的文献中所记载的化合物等。nature395,151(1998)、appl.phys.lett.78,1622(2001)、adv.mater.19,739(2007)、chem.mater.17,3532(2005)、adv.mater.17,1059(2005)、国际公开第2009/100991号、国际公开第2008/101842号、国际公开第2003/040257号、美国专利申请公开第2006/835469号说明书、美国专利申请公开第2006/0202194号说明书、美国专利申请公开第2007/0087321号说明书、美国专利申请公开第2005/0244673号说明书、inorg.chem.40,1704(2001)、chem.mater.16,2480(2004)、adv.mater.16,2003(2004)、angew.chem.lnt.ed.2006,45,7800、appl.phys.lett.86,153505(2005)、chem.lett.34,592(2005)、chem.commun.2906(2005)、inorg.chem.42,1248(2003)、国际公开第2009/050290号、国际公开第2002/015645号、国际公开第2009/000673号、美国专利申请公开第2002/0034656号说明书、美国专利第7332232号说明书、美国专利申请公开第2009/0108737号说明书、美国专利申请公开第2009/0039776号说明书、美国专利第6921915号说明书、美国专利第6687266号说明书、美国专利申请公开第2007/0190359号说明书、美国专利申请公开第2006/0008670号说明书、美国专利申请公开第2009/0165846号说明书、美国专利申请公开第2008/0015355号说明书、美国专利第7250226号说明书、美国专利第7396598号说明书、美国专利申请公开第2006/0263635号说明书、美国专利申请公开第2003/0138657号说明书、美国专利申请公开第2003/0152802号说明书、美国专利第7090928号说明书、angew.chem.lnt.ed.47,1(2008)、chem.mater.18,5119(2006)、inorg.chem.46,4308(2007)、organometallics23,3745(2004)、appl.phys.lett.74,1361(1999)、国际公开第2002/002714号、国际公开第2006/009024号、国际公开第2006/056418号、国际公开第2005/019373号、国际公开第2005/123873号、国际公开第2005/123873号、国际公开第2007/004380号、国际公开第2006/082742号、美国专利申请公开第2006/0251923号说明书、美国专利申请公开第2005/0260441号说明书、美国专利第7393599号说明书、美国专利第7534505号说明书、美国专利第7445855号说明书、美国专利申请公开第2007/0190359号说明书、美国专利申请公开第2008/0297033号说明书、美国专利第7338722号说明书、美国专利申请公开第2002/0134984号说明书、美国专利第7279704号说明书、美国专利申请公开第2006/098120号说明书、美国专利申请公开第2006/103874号说明书、国际公开第2005/076380号、国际公开第2010/032663号、国际公开第2008/140115号、国际公开第2007/052431号、国际公开第2011/134013号、国际公开第2011/157339号、国际公开第2010/086089号、国际公开第2009/113646号、国际公开第2012/020327号、国际公开第2011/051404号、国际公开第2011/004639号、国际公开第2011/073149号、美国专利申请公开第2012/228583号说明书、美国专利申请公开第2012/212126号说明书、日本特开2012-069737号公报、日本特愿2011-181303号公报、日本特开2009-114086号公报、日本特开2003-81988号公报、日本特开2002-302671号公报、日本特开2002-363552号公报等。其中,作为优选的磷光掺杂剂,可举出具有ir作为中心金属的有机金属络合物。进一步优选包含金属-碳键、金属-氮键、金属-氧键、金属-硫键的至少一个配位方式的络合物。(2)主体化合物本发明中使用的主体化合物是在发光层中主要担负电荷的注入及传输的化合物,在有机el元件中实质上没有观测到其自身的发光。主体化合物在发光层所含有的化合物内,该层中的质量比优选为20%以上。主体化合物可以单独使用,或也可以组合使用多种。通过使用多种主体化合物,可以调整电荷的移动,可以使得有机电致发光元件高效率化。以下,对本发明中优选使用的主体化合物进行说明。作为主体化合物,如上所述可以使用本发明中所使用的π共轭类化合物,但没有特别限制。从逆能量移动的观点出发,优选与掺杂剂的激发单态能量相比具有大的激发能量的物质,进一步更优选与掺杂剂的激发三重态能量相比具有大的激发三重态能量的物质。主体化合物在发光层内担负载体的输送及激发子的生成。因此,优选可以在阳离子自由基状态、阴离子自由基状态及激发状态的所有活性种状态下稳定地存在,不引起分解或加成反应等化学变化,另外,优选主体分子不会在通电之后在层中发生埃数量级的移动。另外,特别是在组合使用的发光掺杂剂显示tadf发光的情况下,由于tadf化合物的三重态激发状态的存在时间长,因此,需要如下地进行分子结构的适当设计以使得主体化合物不发生低t1化,即,主体化合物自身的t1能级高、另外主体化合物彼此不会在聚集状态下形成低t1状态、tadf化合物和主体化合物不会形成激基复合物(exciplex)、主体化合物不会由于电场而形成电子异构体等。为了满足这些主要条件,主体化合物自身的电子的跳跃移动性高,且空穴的跳跃移动高,以及形成三重态激发状态时的结构变化小是必须的。作为满足这种主要条件的主体化合物的代表,优选列举咔唑骨架、氮杂咔唑骨架、二苯并呋喃骨架、二苯并噻吩骨架或氮杂二苯并呋喃骨架等具有高t1能级的物质。另外,就主体化合物而言,从具有空穴输送能或电子输送能力、且防止发光的长波长化、以及使有机电致发光元件对于高温驱动时或元件驱动中的发热稳定地工作的观点出发,优选具有高玻璃化转变温度(tg)。优选tg为90℃以上,更优选为120℃以上。在此,玻璃化转变温度(tg)是指使用dsc(differentialscanningcolorimetry:差示扫描量热法),通过根据jisk7121-2012的方法而求出的值。另外,作为用于本发明的主体化合物,如上所述优选使用本发明的π共轭类化合物。这是因为,本发明的π共轭类化合物具有高的t1,对于发光波长短的(即t1及s1的能级高的)发光材料而言,也是可以优选使用的。在本发明的有机el元件中使用公知的主体化合物时,作为其具体例子,可举出以下的文献中记载的化合物等,但本发明并不限定于这些。日本特开2001-257076号公报、日本特开2002-308855号公报、日本特开2001-313179号公报、日本特开2002-319491号公报、日本特开2001-357977号公报、日本特开2002-334786号公报、日本特开2002-8860号公报、日本特开2002-334787号公报、日本特开2002-15871号公报、日本特开2002-334788号公报、日本特开2002-43056号公报、日本特开2002-334789号公报、日本特开2002-75645号公报、日本特开2002-338579号公报、日本特开2002-105445号公报、日本特开2002-343568号公报、日本特开2002-141173号公报、日本特开2002-352957号公报、日本特开2002-203683号公报、日本特开2002-363227号公报、日本特开2002-231453号公报、日本特开2003-3165号公报、日本特开2002-234888号公报、日本特开2003-27048号公报、日本特开2002-255934号公报、日本特开2002-260861号公报、日本特开2002-280183号公报、日本特开2002-299060号公报、日本特开2002-302516号公报、日本特开2002-305083号公报、日本特开2002-305084号公报、日本特开2002-308837号公报、美国专利申请公开第2003/0175553号说明书、美国专利申请公开第2006/0280965号说明书、美国专利申请公开第2005/0112407号说明书、美国专利申请公开第2009/0017330号说明书、美国专利申请公开第2009/0030202号说明书、美国专利申请公开第2005/0238919号说明书、国际公开第2001/039234号、国际公开第2009/021126号、国际公开第2008/056746号、国际公开第2004/093207号、国际公开第2005/089025号、国际公开第2007/063796号、国际公开第2007/063754号、国际公开第2004/107822号、国际公开第2005/030900号、国际公开第2006/114966号、国际公开第2009/086028号、国际公开第2009/003898号、国际公开第2012/023947号、日本特开2008-074939号公报、日本特开2007-254297号公报、欧州专利第2034538号说明书、国际公开第2011/055933号、国际公开第2012/035853号、日本特开2015-38941号公报等。在日本特开2015-38941号公报中记载的主体化合物的例子包括:说明书的[0255]~[0293]中记载的化合物h-1~h-230。以下,作为用于本发明的主体化合物,示出具体的化合物例子,但并不限定于这些。[化学式96]另外,用于本发明的主体化合物优选为下述通式(i)所表示的化合物。下述通式(i)所表示的化合物具有双极性,且非常稳定。因此,主体化合物为下述通式(i)所表示的化合物时,有机el元件的寿命容易变长。[化学式97]通式(i)上述通式(i)中,x101表示nr101、氧原子、硫原子、亚硫酰基、硫酰基、cr102r103或sir104r105。另外,y1~y8分别独立地表示cr106、或氮原子。在此,r101~r106分别独立地表示氢原子或取代基,可以相互键合而形成环。可以为r101~r106的取代基的例子包括以下基团。直链状或支链状的烷基(例如甲基、乙基、丙基、异丙基、叔丁基、戊基、己基、辛基、十二烷基、十三烷基、十四烷基、十五烷基等);烯基(例如乙烯基、烯丙基等);炔基(例如乙炔基、丙炔基等);芳香族烃环基(也称为芳香族碳环基或芳基等。例如,从苯环、联苯、萘环、薁环、蒽环、菲环、芘环、环、并四苯环、二苯并萘(triphenylene)环、邻联三苯环、间联三苯环、对联三苯环、苊环、晕苯环、茚环、芴环、荧蒽环、二苯并萘(triphenylene)环、并五苯环、苝环、戊芬环、苉环、芘环、吡蒽环、蒽嵌蒽环、四氢化萘等中所衍生出的基团);芳香族杂环基(例如呋喃环、二苯并呋喃环、噻吩环、二苯并噻吩环、噁唑环、吡咯环、吡啶环、哒嗪环、嘧啶环、吡嗪环、三嗪环、苯并咪唑环、噁二唑环、三唑环、咪唑环、吡唑环、噻唑环、吲哚环、吲唑环、苯并咪唑环、苯并噻唑环、苯并噁唑环、喹喔啉环、喹唑啉环、邻二氮杂萘环、喹啉环、异喹啉环、酞嗪环、1,5-二氮杂萘环、咔唑环、咔啉环、二氮杂咔唑环(从构成咔啉环的烃环的碳原子中的一个进一步被氮原子取代而成的环等所衍生出的基团。另外,有时将咔啉环和二氮杂咔唑环合称为“氮杂咔唑环”。);非芳香族烃环基(例如环戊基、环己基等);非芳香族杂环基(例如吡咯烷基、咪唑烷基、吗啉基、噁唑烷基等);烷氧基(例如甲氧基、乙氧基、丙氧基、戊氧基、己氧基、辛氧基、十二烷氧基等);环烷氧基(例如环戊氧基、环己氧基等);芳氧基(例如苯氧基、萘氧基等);烷基硫基(例如甲基硫基、乙基硫基、丙基硫基、戊基硫基、己基硫基、辛基硫基、十二烷基硫基等);环烷基硫基(例如环戊基硫基、环己基硫基等);芳基硫基(例如苯基硫基、萘基硫基等):烷氧基羰基(例如甲氧基羰基、乙氧基羰基、丁氧基羰基、辛氧基羰基、十二烷氧基羰基等);芳氧基羰基(例如苯氧基羰基、萘氧基羰基等);氨磺酰基(例如氨基磺酰基、甲基氨基磺酰基、二甲基氨基磺酰基、丁基氨基磺酰基、己基氨基磺酰基、环己基氨基磺酰基、辛基氨基磺酰基、十二烷基氨基磺酰基、苯基氨基磺酰基、萘基氨基磺酰基、2-吡啶氨基磺酰基等);酰基(例如乙酰基、乙基羰基、丙基羰基、戊基羰基、环己基羰基、辛基羰基、2-乙基己基羰基、十二烷基羰基、苯基羰基、萘基羰基、吡啶羰基等);酰氧基(例如乙酰氧基、乙基羰氧基、丁基羰氧基、辛基羰氧基、十二烷基羰氧基、苯基羰氧基等);酰胺基(例如甲基羰基氨基、乙基羰基氨基、二甲基羰基氨基、丙基羰基氨基、戊基羰基氨基、环己基羰基氨基、2-乙基己基羰基氨基、辛基羰基氨基、十二烷基羰基氨基、苯基羰基氨基、萘基羰基氨基等);氨基甲酰基(例如氨基羰基、甲基氨基羰基、二甲基氨基羰基、丙基氨基羰基、戊基氨基羰基、环己基氨基羰基、辛基氨基羰基、2-乙基己基氨基羰基、十二烷基氨基羰基、苯基氨基羰基、萘基氨基羰基、2-吡啶氨基羰基等);酰脲基(例如甲基酰脲基、乙基酰脲基、戊基酰脲基、环己基酰脲基、辛基酰脲基、十二烷基酰脲基、苯基酰脲基、萘基酰脲基、2-吡啶氨基酰脲基等);亚硫酰基(例如甲基亚硫酰基、乙基亚硫酰基、丁基亚硫酰基、环己基亚硫酰基、2-乙基己基亚硫酰基、十二烷基亚硫酰基、苯基亚硫酰基、萘基亚硫酰基、2-吡啶亚硫酰基等);烷基磺酰基(例如甲基磺酰基、乙基磺酰基、丁基磺酰基、环己基磺酰基、2-乙基己基磺酰基、十二烷基磺酰基等);芳基磺酰基或杂芳基磺酰基(例如苯基磺酰基、萘基磺酰基、2-吡啶磺酰基等);氨基(例如氨基、乙基氨基、二甲基氨基、丁基氨基、环戊基氨基、2-乙基己基氨基、十二烷基氨基、苯胺基、萘基氨基、2-吡啶氨基、二苯基氨基等);卤原子(例如氟原子、氯原子、溴原子等);氟化烃基(例如氟甲基、三氟甲基、五氟乙基、五氟苯基等);氰基;硝基;羟基;硫醇基;甲硅烷基(例如三甲基甲硅烷基、三异丙基甲硅烷基、三苯基甲硅烷基、苯基二乙基甲硅烷基等);氘原子。上述中,作为优选的基团,可举出:直链或支链烷基、芳香族烃环基、芳香族杂环基、氨基、氟原子、氟化烃基、氰基、及甲硅烷基。另外,ar101及ar102分别独立地表示任选被取代的芳基、或任选被取代的杂芳基。“任选被取代的芳基”中的芳基优选为从碳原子数6~24的芳香族烃环衍生出的基团。这种芳香族烃环的例子包括:苯环、茚环、萘环、薁环、芴环、菲环、蒽环、苊烯环、联苯撑环、并四苯环、芘环、戊搭烯环、醋蒽烯环、庚搭烯环、二苯并萘(triphenylene)环、as-苯并二茚环、环、s-苯并二茚环、七曜烯环、1,8-苯嵌萘(phenalene)环、荧蒽环、苝环、醋菲烯环、联苯环、联三苯环及联四苯环等。优选为苯环、联苯环、联三苯环。另外,“任选被取代的杂芳基”中的杂芳基优选为从各种芳香族杂环衍生出的基团。这种芳香族杂环的例子包括:吡咯环、吲哚环、咔唑环、吲哚并吲哚环、9,10-二氢吖啶环、5,10-二氢吩嗪环、吩噁嗪环、吩噻嗪环、二苯并噻吩环、苯并呋喃基吲哚环、苯并噻吩并吲哚环、吲哚并咔唑环、苯并呋喃基咔唑环、苯并噻吩并咔唑环、苯并噻吩并苯并噻吩环、苯并咔唑环、二苯并咔唑环、氮杂咔唑环、氮杂二苯并呋喃环、二氮杂咔唑环、二氮杂二苯并呋喃环、二苯并噻吩氧化物环、二苯并噻吩二氧化物环、吡啶环、哒嗪环、嘧啶环、吡嗪环、三嗪环、喹啉环、异喹啉环、喹唑啉环、邻二氮杂萘环、喹喔啉环、酞嗪环、蝶啶环、邻二氮杂菲环、菲绕啉环、二苯并呋喃环、二苯并噻咯环、二苯并硼杂环戊二烯环、二苯并磷杂环戊二烯氧化物环等。优选为咔唑环、苯并咔唑环、氮杂咔唑环、氮杂二苯并呋喃环、二氮杂咔唑环、二氮杂二苯并呋喃环、吡啶环、嘧啶环、三嗪环。芳基或杂芳基可具有的取代基的例子包括:任选被烷基取代的芳基、氟原子、氰基、任选被氟取代的烷基、任选被取代的羰基、任选被取代的硫酰基、任选被取代的氧化膦基、任选被取代的硼烷基、任选被取代的杂环基、任选被取代的氨基、任选被取代的甲硅烷基等。上述“烷基”可以为直链、支链或环状的任一种,例如可以为碳原子数1~20的直链或支链的烷基、或碳原子数5~20的环状的烷基。烷基例子包括:甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、环己基、2-乙基己基、正庚基、正辛基、2-己基辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基、及正二十基等。另外,“芳基”及“杂环基”可以与ar101及ar102所表示的芳基或杂芳基含义相同。另外,上述通式(i)中,n101及n102分别表示0~4的整数,但r101为氢原子的情况下,n101表示1~4的整数。具有上述通式(i)所表示的结构的主体化合物更优选具有下述通式(ii)所表示的结构。[化学式98]通式(ii)通式(ii)中,x101表示nr101、氧原子、硫原子、亚硫酰基、硫酰基、cr102r103或sir104r105。r101~r105分别独立地表示氢原子或取代基,可以相互键合而形成环。需要说明的是,r101~r105与通式(i)的r101~r105意义相同。ar101及ar102分别独立地表示任选被取代的芳基、任选被取代的杂芳基,ar101及ar102与通式(i)的ar101及ar102意义相同。另外,n102表示0~4的整数。另外,通式(ii)所表示的化合物优选为下述通式(iii-1)、(iii-2)或(iii-3)所表示的化合物。[化学式99]通式(iii-1)~(iii-3)中,x101、ar102、n102与所述通式(ii)中的x101、ar102、n102意义相同。另外,通式(iii-2)中,r104与所述通式(i)中的r104意义相同。通式(iii-1)~(iii-3)中,含有x101而形成的稠环、咔唑环及苯环可以在不阻碍主体化合物的功能的范围内进一步具有取代基。可举出上述通式(i)、(ii)、或(iii-1)~(iii-3)所表示的主体化合物的优选的具体例,但这些化合物有时进一步具有取代基,或存在结构异构体等,并不限定于本记载。[化学式100][化学式101][化学式102][化学式103][化学式104][化学式105][化学式106][化学式107][化学式108][化学式109][化学式110][化学式111][化学式112][化学式113][化学式114][化学式115][化学式116][化学式117]《电子传输层》本发明中电子传输层由具有传输电子的功能的材料构成,只要具有将从阴极注入的电子传递至发光层的功能即可。对本发明的电子传输层的总层厚没有特别限制,通常为2nm~5μm的范围,更优选为2~500nm,进一步优选为5~200nm。另外,在有机el元件中,从电极导出发光层中产生的光时,已知从发光层中直接导出的光、以及被导出光的电极和位于对极的电极反射后导出的光会发生干涉。光在阴极中被反射的情况下,通过在数nm~数μm之间适当调整电子传输层的总层厚,可以有效地利用该干涉效果。另一方面,使电子传输层的层厚变厚时,电压容易上升,因此,特别是在层厚很厚的情况下,电子传输层的电子移动度优选为10-5cm2/v·s以上。作为电子传输层所使用的材料(以下,称为电子传输材料),只要具有电子的注入性或传输性、空穴的屏障性的任一种即可,可以从现有公知的化合物中选择任意的物质来使用。可列举例如:含氮芳香族杂环衍生物(咔唑衍生物、氮杂咔唑衍生物(构成咔唑环的碳原子的一个以上被氮原子取代而成的化合物)、吡啶衍生物、嘧啶衍生物、吡嗪衍生物、哒嗪衍生物、三嗪衍生物、喹啉衍生物、喹喔啉衍生物、菲绕啉衍生物、氮杂三邻亚苯衍生物、噁唑衍生物、噻唑衍生物、噁二唑衍生物、噻二唑衍生物、三唑衍生物、苯并咪唑衍生物、苯并噁唑衍生物、苯并噻唑衍生物等)、二苯并呋喃衍生物、二苯并噻吩衍生物、噻咯衍生物、芳香族烃环衍生物(萘衍生物、蒽衍生物、三邻亚苯衍生物等)等。另外,在配体上具有羟基喹啉骨架或二苯并羟基喹啉骨架的金属络合物、例如三(8-羟基喹啉)铝(alq)、三(5,7-二氯-8-羟基喹啉)铝、三(5,7-二溴-8-羟基喹啉)铝、三(2-甲基-8-羟基喹啉)铝、三(5-甲基-8-羟基喹啉)铝、双(8-羟基喹啉)锌(znq)等、及这些金属络合物的中心金属取代为in、mg、cu、ca、sn、ga或pb而成的金属络合物也可以作为电子传输材料使用。此外,无金属或金属酞菁、或它们的末端用烷基或磺酸基等取代而成的物质也可以作为电子传输材料优选使用。另外,作为发光层的材料列举的二苯乙烯基吡嗪衍生物也可以作为电子传输材料使用,与空穴注入层、空穴传输层同样地,n型-si、n型-sic等无机半导体也可以作为电子传输材料使用。另外,也可以使用在高分子链上导入这些材料而成的高分子材料、或将这些材料作为高分子的主链而得到的高分子材料。在本发明的电子传输层中,在电子传输层中将掺杂材料作为客体材料进行掺杂,可以形成n性高的(富电子)电子传输层。作为掺杂材料,可列举金属络合物或卤化金属等金属化合物等n型掺杂剂。作为这种构成的电子传输层的具体例,可列举例如日本特开平4-297076号公报、日本特开平10-270172号公报、日本特开2000-196140号公报、日本特开2001-102175号公报、j.appl.phys.,95,5773(2004)等文献所记载的电子传输层。作为本发明的有机el元件所使用的、公知的优选的电子传输材料的具体例,可列举以下的文献中记载的化合物等,但本发明并不限定于这些。美国专利第6528187号说明书、美国专利第7230107号说明书、美国专利申请公开第2005/0025993号说明书、美国专利申请公开第2004/0036077号说明书、美国专利申请公开第2009/0115316号说明书、美国专利申请公开第2009/0101870号说明书、美国专利申请公开第2009/0179554号说明书、国际公开第2003/060956号、国际公开第2008/132085号、appl.phys.lett.75,4(1999)、appl.phys.lett.79,449(2001)、appl.phys.lett.81,162(2002)、appl.phys.lett.81,162(2002)、appl.phys.lett.79,156(2001)、美国专利第7964293号说明书、美国专利申请公开第2009/030202号说明书、国际公开第2004/080975号、国际公开第2004/063159号、国际公开第2005/085387号、国际公开第2006/067931号、国际公开第2007/086552号、国际公开第2008/114690号、国际公开第2009/069442号、国际公开第2009/066779号、国际公开第2009/054253号、国际公开第2011/086935号、国际公开第2010/150593号、国际公开第2010/047707号、欧洲专利2311826号、日本特开2010-251675号公报、日本特开2009-209133号公报、日本特开2009-124114号公报、日本特开2008-277810号公报、日本特开2006-156445号公报、日本特开2005-340122号公报、日本特开2003-45662号公报、日本特开2003-31367号公报、日本特开2003-282270号公报、国际公开第2012/115034号等。作为本发明中的更优选公知的电子传输材料,可列举含有至少一个氮原子的芳香族杂环化合物或含有磷原子的化合物,可列举例如:吡啶衍生物、嘧啶衍生物、吡嗪衍生物、三嗪衍生物、二苯并呋喃衍生物、二苯并噻吩衍生物、氮杂二苯并呋喃衍生物、氮杂二苯并噻吩衍生物、咔唑衍生物、氮杂咔唑衍生物、苯并咪唑衍生物、芳基氧化膦等。电子传输材料可以单独使用,另外也可以组合使用多种。《空穴阻挡层》空穴阻挡层广义上为具有电子传输层功能的层,优选由具有传输电子的功能、并且传输空穴的能力小的材料构成,可以通过一边传输电子、一边阻挡空穴而提高电子和空穴的再键合概率。另外,可以将上述的电子传输层的构成根据需要作为本发明的空穴阻挡层使用。设置于本发明的有机el元件的空穴阻挡层优选与发光层的阴极侧邻接而设置。作为本发明中空穴阻挡层的层厚,优选为3~100nm的范围,进一步优选为5~30nm的范围。作为空穴阻挡层所使用的材料,优选使用上述的电子传输层所使用的材料,另外,用作上述的主体化合物的材料也优选用于空穴阻挡层。《电子注入层》本发明的电子注入层(也称为“阴极缓冲层”)是指为了降低驱动电压或提高发光亮度而设置于阴极和发光层之间的层,详细地记载于“有机el元件和其工业化最前沿(1998年11月30日nts社发行)”的第2编第2章“电极材料”(123~166页)。本发明中电子注入层根据需要而设置,如上所述可以存在于阴极和发光层之间、或阴极和电子传输层之间。电子注入层优选为极薄的膜,层厚因原料的不同而不同,但该层厚优选为0.1~5nm的范围。另外,也可以为构成材料间断地存在的不均匀的层(膜)。电子注入层在日本特开平6-325871号公报、日本特开平9-17574号公报、日本特开平10-74586号公报等中也记载有其详细,作为优选用于电子注入层的材料的具体例,可列举以锶或铝等为代表的金属、以氟化锂、氟化钠、氟化钾等为代表的碱金属化合物、以氟化镁、氟化钙等为代表的碱土金属化合物、以氧化铝为代表的金属氧化物、以8-羟基喹啉基锂(liq)等为代表的金属络合物等。另外,也可以使用上述的电子传输材料。另外,上述的电子注入层所使用的材料可以单独使用,也可以组合使用多种。《空穴传输层》本发明中空穴传输层由具有传输空穴的功能的材料构成,只要具有将从阳极注入的空穴传递至发光层的功能即可。对本发明的空穴传输层的总层厚没有特别限制,通常为5nm~5μm的范围,更优选为2~500nm,进一步优选为5~200nm。作为空穴传输层所使用的材料(以下,称为空穴传输材料),只要具有空穴的注入性或传输性、电子的屏障性的任一种即可,可以从现有公知的化合物中选择任意的物质而使用。可列举例如:卟啉衍生物、酞菁衍生物、噁唑衍生物、噁二唑衍生物、三唑衍生物、咪唑衍生物、吡唑啉衍生物、吡唑啉酮衍生物、亚苯基二胺衍生物、腙衍生物、茋衍生物、聚芳基链烷衍生物、三芳基胺衍生物、咔唑衍生物、吲哚并咔唑衍生物、异吲哚衍生物、蒽或萘等并苯系衍生物、芴衍生物、芴酮衍生物、及聚乙烯基咔唑、在主链或侧链上导入有芳香族胺的高分子材料或低聚物、聚硅烷、导电性聚合物或低聚物(例如pedot/pss、苯胺系共聚物、聚苯胺、聚噻吩等)等。作为三芳基胺衍生物,可列举:以α-npd(4,4′-双[n-(1-萘基)-n-苯基氨基]联苯)为代表的联苯胺型、或以mtdata为代表的星爆型、在三芳基胺连结芯部中具有芴或蒽的化合物等。另外,日本特表2003-519432号公报或日本特开2006-135145号公报等中所记载的六氮杂三邻亚苯衍生物也同样地可以作为空穴传输材料使用。另外还可以使用掺杂了杂质的p性高的空穴传输层。作为其实例,可列举:日本特开平4-297076号公报、日本特开2000-196140号公报、日本特开2001-102175号公报的各公报、j.appl.phys.,95,5773(2004)等中所记载的空穴传输层。另外,也可以使用日本特开平11-251067号公报、j.huanget.al.著文献(appliedphysicsletters80(2002),p.139)中所记载的所谓p型空穴传输材料或p型-si、p型-sic等无机化合物。另外,还优选使用在以ir(ppy)3为代表的中心金属具有ir或pt的邻金属化有机金属络合物。作为空穴传输材料,可以使用上述的物质,但优选使用三芳基胺衍生物、咔唑衍生物、吲哚并咔唑衍生物、氮杂三邻亚苯衍生物、有机金属络合物、在主链或侧链上导入有芳香族胺的高分子材料或低聚物等。作为本发明的有机el元件所使用的、公知的优选的空穴传输材料的具体例,除上述中列举的文献之外,可列举以下的文献中记载的化合物等,但本发明并不限定于这些。例如为:appl.phys.lett.69,2160(1996)、j.lumin.72-74,985(1997)、appl.phys.lett.78,673(2001)、appl.phys.lett.90,183503(2007)、appl.phys.lett.51,913(1987)、synth.met.87,171(1997)、synth.met.91,209(1997)、synth.met.111,421(2000)、sidsymposiumdigest,37,923(2006)、j.mater.chem.3,319(1993)、adv.mater.6,677(1994)、chem.mater.15,3148(2003)、美国专利申请公开第2003/0162053号说明书、美国专利申请公开第2002/0158242号说明书、美国专利申请公开第2006/0240279号说明书、美国专利申请公开第2008/0220265号说明书、美国专利第5061569号说明书、国际公开第2007/002683号、国际公开第2009/018009号、ep650955、美国专利申请公开第2008/0124572号说明书、美国专利申请公开第2007/0278938号说明书、美国专利申请公开第2008/0106190号说明书、美国专利申请公开第2008/0018221号说明书、国际公开第2012/115034号、日本特表2003-519432号公报、日本特开2006-135145号公报、美国专利申请号13/585981号等。空穴传输材料可以单独使用,另外也可以组合使用多种。《电子阻挡层》电子阻挡层广义上是指具有空穴传输层的功能的层,优选由具有传输空穴的功能且传输电子的能力小的材料构成,可以通过一边传输空穴、一边阻挡电子而提高电子和空穴的再键合概率。另外,根据需要可以将上述的空穴传输层的构成作为本发明中的电子阻挡层使用。设置于本发明的有机el元件的电子阻挡层优选与发光层的阳极侧邻接而设置。作为本发明的电子阻挡层的层厚,优选为3~100nm的范围内,进一步优选为5~30nm的范围内。作为电子阻挡层所使用的材料,优选使用上述的空穴传输层所使用的材料,另外,上述的主体化合物还优选用于电子阻挡层。《空穴注入层》本发明的空穴注入层(也称为“阳极缓冲层”)是指为了降低驱动电压或提高发光亮度而设置于阳极和发光层之间的层,详细地记载于“有机el元件和其工业化最前沿(1998年11月30日nts社发行)”的第2编第2章“电极材料”(123~166页)。本发明中空穴注入层根据需要而设置,如上所述可以存在于阳极和发光层或阳极和空穴传输层之间。空穴注入层在日本特开平9-45479号公报、日本特开平9-260062号公报、日本特开平8-288069号公报等中也记载有其详细,作为空穴注入层所使用的材料,可列举例如上述的空穴传输层所使用的材料等。其中,优选以铜酞菁为代表的酞菁衍生物、日本特表2003-519432号公报或日本特开2006-135145号公报等所记载的六氮杂三邻亚苯衍生物、以氧化钒为代表的金属氧化物、非晶碳、聚苯胺(人造绿宝石)或聚噻吩等导电性高分子、以三(2-苯基吡啶)铱络合物等为代表的邻金属化络合物、三芳基胺衍生物等。上述的空穴注入层所使用的材料可以单独使用,另外也可以组合使用多种。《添加物》上述的本发明的各层可以进一步含有其它添加物。作为添加物,可列举例如溴、碘及氯等卤元素或卤化化合物、pd、ca、na等碱金属或碱土金属、过渡金属的化合物或络合物、盐等。添加物的含量可以任意地确定,相对于所含有的层的总质量%,优选为1000ppm以下,更优选为500ppm以下,进一步优选为50ppm以下。其中,根据提高电子或空穴的传输性的目的或有利于激发子的能量移动的目的等,有时也不在该范围内。《各层的形成方法》对上述的各层(空穴注入层、空穴传输层、发光层、空穴阻挡层、电子传输层、电子注入层、中间层等)的形成方法进行说明。上述的各层的形成方法没有特别限制,可以使用现有公知的例如真空蒸镀法、湿式法(也称为湿工艺)等的形成方法。作为湿式法,有旋涂法、流延法、喷墨法、印刷法、点胶涂敷法(dyecoating)、刮板涂布法、辊涂法、喷涂法、流帘涂布法、lb法(langmuir-blodgett法)等,但从容易得到均质的薄膜、且高生产率方面考虑,优选点胶涂敷法、辊涂法、喷墨法、喷涂法等辊对辊方式适性高的方法。作为将本发明所使用的有机el材料进行溶解或分散的液体介质,例如可以使用甲基乙基酮、环己酮等酮类、醋酸乙酯等脂肪酸酯类、二氯苯等卤化烃类、甲苯、二甲苯、均三甲苯、环己基苯等芳香族烃类、环己烷、十氢化萘、十二烷等脂肪族烃类、dmf、dmso等有机溶剂。另外,作为分散方法,可以通过超声波、高剪切力分散或介质分散等分散方法进行分散。另外,每层可以使用不同的成膜法。在成膜中采用蒸镀法的情况下,其蒸镀条件因使用的化合物的种类等而不同,一般而言,优选在舟加热温度50~450℃、真空度1×10-6~1×10-2pa、蒸镀速度0.01~50nm/秒、基板温度-50~300℃、层(膜)厚0.1nm~5μm、优选5~200nm的范围内适当选择。各层的形成优选通过一次抽真空连贯地将空穴注入层直至阴极制成,但也可以在中途取出并实施不同的成膜法。此时优选在干燥非活性气体气氛下进行操作。《阳极》作为有机el元件中的阳极,优选使用将工作函数大(4ev以上、优选4.5ev以上)的金属、合金、导电性化合物及它们的混合物作为电极物质的阳极。作为这种电极物质的具体例,可列举au等金属、cui、铟锡氧化物(ito)、sno2、zno等导电性透明材料。另外,可以使用idixo(in2o3-zno)等非晶质且可制作透明导电膜的材料。就阳极而言,使这些电极物质通过蒸镀或溅射等方法而形成薄膜,可以用光刻法形成所期望的形状的图案,或不太要求图案精度的情况(100μm以上左右)下,可以在上述电极物质的蒸镀或溅射时经由所期望的形状的掩模而形成图案。或者,在使用像有机导电性化合物那样可涂布的物质的情况下,也可以使用印刷方式、涂敷方式等湿式成膜法。在从该阳极导出发光的情况下,优选使透过率大于10%,另外,作为阳极的片材电阻优选为数百ω/□以下。阳极的膜厚也因材料的不同而不同,通常在10nm~1μm、优选10~200nm的范围内选择。《阴极》作为阴极,可使用将工作函数小(4ev以下)的金属(称为电子注入性金属)、合金、导电性化合物及它们的混合物作为电极物质的阴极。作为这种电极物质的具体例,可列举:钠、钠-钾合金、镁、锂、镁/铜混合物、镁/银混合物、镁/铝混合物、镁/铟混合物、铝/氧化铝(al2o3)混合物、铟、锂/铝混合物、铝、稀土金属等。其中,从相对于电子注入性及氧化等的耐久性方面考虑,优选电子注入性金属和工作函数的值比其大且稳定的金属即第二金属的混合物、例如、镁/银混合物、镁/铝混合物、镁/铟混合物、铝/氧化铝(al2o3)混合物、锂/铝混合物、铝等。阴极可以通过蒸镀或溅射等方法使这些电极物质形成薄膜来制作。另外,作为阴极的片材电阻优选为数百ω/□以下,膜厚通常在10nm~5μm、优选50~200nm的范围内选择。需要说明的是,为了使发光的光透过,只要有机el元件的阳极或阴极中的任一个为透明或半透明,则发光亮度提高且合适。另外,在阴极上以1~20nm的膜厚制作上述金属之后,通过阳极的说明中所列举的在其上制作导电性透明材料,可以制作透明或半透明的阴极,可以使用该方法来制作阳极和阴极这两者都具有透过性的元件。《使用有特定的π共轭类化合物的有机el元件》本发明的π共轭类化合物中,上述通式(201)所表示的特定的π共轭类化合物即使在发光层中单独存在,也具有高的发光效率,因此,目前需要的主体化合物变得不需要。因此,根据该特定的π共轭类化合物,可以使有机el元件的结构变得简单而不降低发光效率和耐久性。以下,对使用有特定的π共轭类化合物的有机el元件进行说明。使用了特定的π共轭类化合物的有机el元件包含阳极、阴极、和配置于它们之间的有机层。有机层实质上不含有通式(201)所表示的π共轭类化合物以外的主体化合物。即,有机层优选实质上由通式(201)所表示的π共轭类化合物构成(第1方式)、或实质上由通式(201)所表示的π共轭类化合物和客体化合物构成(第2方式)。在第1有机el元件中,通式(201)所表示的π共轭类化合物作为主体化合物兼发光性化合物起作用,在第2有机el元件中,通式(201)所表示的π共轭类化合物可作为主体化合物起作用。“有机层实质上由通式(201)所表示的π共轭类化合物构成”是指:有机层可以含有对发光性能和耐久性能不产生影响的程度的杂质,具体而言,是指有机层中的通式(201)所表示的π共轭类化合物的含量为95体积%以上,优选为99体积%以上,特别优选为100体积%。同样地,“实质上由通式(201)所表示的π共轭类化合物和客体化合物构成”是指:有机层中的通式(201)所表示的π共轭类化合物和客体化合物的含量的合计为95体积%以上,优选为99体积%以上,特别优选为100体积%。优选有机层为发光层。发光层可以由单层构成,也可以由多层构成。这种发光层即使不含有以往的主体化合物,也可以以高效率发光。因此,可以使有机el元件的构成简单,因此,可以提高生产率或可靠性。以下,对各自的方式进行说明。(关于第1方式的有机el元件)图5a~5c是表示使用了特定的π共轭类化合物的第1方式的有机el元件的构成例的图。如图5a~5c所示,第1方式的有机el元件1包含设置于支撑基板2上的阳极3、阴极4、和配置于它们之间的发光层5。发光层5实质上由通式(201)所表示的π共轭类化合物构成。与上述的有机el元件同样,发光层5是提供从电极(有机el元件含有其它层的情况下,为与之相邻的层)注入的电子及空穴进行再结合经由激发子而发光的场所的层。发光的部分可以为发光层的层内,有机el元件含有其它层的情况下,也可以是发光层和预支相邻的层的界面。发光层5可以由单层构成,也可以由多层构成。发光层5的层厚的总和、或发光层5每1层的层厚可以与上述有机el元件同样。在此,发光层5中所含的通式(201)所表示的π共轭类化合物优选具有tadf性。这是因为,由于通过电场激发而生成的75%的t1能级向s1进行项间跃迁而进行荧光发光,因此,可以没有损失地使用生成的激发能量,容易得到高的外部量子收率。有机el元件含有其它层的情况下,与之相邻的层中所含的化合物和发光层5中所含的通式(201)所表示的π共轭类化合物可以形成激发络合物而进行发光。第1方式的有机el元件1可以根据需要还含有配置于发光层5和阳极3之间的空穴传输层6、及配置于发光层5和阴极4之间的电子传输层7的至少一个(参照图5b)。空穴传输层6或电子传输层7分别作为调整空穴或电子的移动度的层起作用,提高再结合概率而提高发光效率,或可以使驱动电压降低。第1方式的有机el元件1可以根据需要还含有配置于发光层5和阳极3之间的空穴注入层8、及配置于发光层5和阴极4之间的电子注入层9的至少一个(参照图5c)。空穴注入层8或电子注入层9可以提高发光层5和由无机物构成的阳极3或阴极4的接合性,可以容易将电子或空穴注入于有机层。第1方式的有机el元件1可以根据需要还含有配置于发光层5和阴极4之间的空穴阻挡层(未图示)、及配置于发光层5和阳极3之间的电子阻挡层(不图示)的至少一个。其中,从减少构成有机el元件的层的数、容易提高生产率或可靠性的观点出发,第1方式的有机el元件优选在发光层5和阳极3之间、及在发光层5和阴极4之间不含有其它层(参照图5a)。即,优选发光层5的一个面与阳极3相接,且发光层5的另一个面与阴极4相接。作为第1方式的有机el元件中的代表的元件结构,可以采用与上述的有机el元件的结构相同的结构,但并不限定于上述的结构。该有机el元件中所含的电子传输层、空穴阻挡层、电子注入层、空穴传输层、电子阻挡层、空穴注入层、或这些中所含的添加物、各层的形成方法可以采用与上述有机el元件相同的结构。(关于第2方式的有机el元件)图6是表示使用有特定的π共轭类化合物的第2方式的有机el元件的构成例的图。如图6所示,第2方式的有机el元件1’包含设置于支撑基板2上的阳极3、阴极4、和一个面与阳极3相接、且另一个面与阴极4相接的发光层5’。发光层5’实质上由通式(201)所表示的π共轭类化合物和客体化合物构成。第1方式的有机el元件中的发光层5同样,发光层5’可以由单层构成,也可以由多层构成。发光层5’的层厚的总和及每1层的层厚可以与第1方式的有机el元件中的发光层5的层厚的总和及每1层的层厚同样。发光层5’中所含的客体化合物可以为通式(201)所表示的π共轭类化合物以外的荧光发光性化合物(也称为荧光发光性掺杂剂、或荧光掺杂剂)或磷光发光性化合物(也称为磷光发光性掺杂剂、或磷光掺杂剂)。客体化合物可以组合使用多种,也可以组合结构不同的荧光发光性化合物、或组合荧光发光性化合物和磷光发光性化合物。由此,可以得到任意的发光色。发光层5’中的客体化合物的含量优选为发光层5’的0.1~20体积%,更优选为0.1~5体积%,进一步优选为0.1~1体积%。这是因为,客体化合物的含量超过20体积%时,发光层5’中的电子和空穴的再结合容易在客体化合物上产生,在客体化合物中进行再结合而以75%的概率生成的t1能量不进行发光而是发生热失活,成为降低外部量子效率的主要原因。客体化合物可以在发光层5’的层厚方向上以均匀浓度存在,也可以具有任意的浓度分布。发光层5’中所含的通式(201)所表示的π共轭类化合物优选具有tadf性。通式(201)所表示的π共轭类化合物具有tadf性时,在该π共轭类化合物上生成的t1能量向s1进行逆系间穿越。而且,进一步向客体化合物上进行能量移动(荧光共振能量移动(fret)),客体化合物发光,因此,可以得到高的量子收率。因此,由通式(201)所表示的π共轭类化合物和客体化合物这2种成分构成的发光层5’中,该π共轭类化合物的s1和t1的能级优选比客体化合物的s1和t1的能级高。图4表示通式(201)所表示的π共轭类化合物作为主体化合物起作用时的示意图。图4为一例,在通式(201)所表示的π共轭类化合物上生成的三重态激发子的生成过程并不仅限于电场激发,也包含来自发光层内或周边层界面的能量移动或电子移动等。另外,图4中,使用荧光发光性化合物作为客体化合物(发光性化合物)来表示,但并不限定于此,可以使用磷光发光性化合物,也可以使用荧光发光性化合物和磷光发光性化合物这两者。通式(201)所表示的π共轭类化合物作为主体化合物起作用时,优选通式(201)所表示的π共轭类化合物的发光光谱和客体化合物的吸收光谱重叠。发光层5’中,通式(201)所表示的π共轭类化合物和客体化合物可以在激发状态下形成络合物(形成激基复合物)而发光。本发明的有机el元件和用于本发明的化合物的发光的颜色可以采用与上述的有机el元件同样的颜色。可在第2方式的发光层中使用的发光性掺杂剂或磷光发光性掺杂剂可以采用与能够在上述有机el元件中使用的荧光发光性掺杂剂或磷光性掺杂剂同样的掺杂剂。需要说明的是,作为荧光发光性掺杂剂,可以选择延迟荧光性掺杂剂来使用。延迟荧光性掺杂剂是通式(201)所表示的π共轭类化合物以外的δest为0.5ev以下的化合物。发光层5’可用与上述有机el元件的各层的形成方法同样的方法形成。[支撑基板]作为可以用于本发明的有机el元件的支撑基板(以下,也称为基板、基材等),对玻璃、塑料等种类没有特别限定,另外,可以为透明,也可以为不透明。在从支撑基板侧取出光的情况下,支撑基板优选为透明。作为优选使用的透明的支撑基板,可举出玻璃、石英、透明树脂膜。特别优选支撑基板为可以对有机el元件赋予挠性的树脂膜。作为树脂膜,可列举例如:聚对苯二甲酸乙二醇酯(pet)、聚萘二甲酸乙二醇酯(pen)等聚酯、聚乙烯、聚丙烯、玻璃纸、纤维素二乙酸酯、纤维素三乙酸酯(tac)、乙酸丁酸纤维素、乙酸丙酸纤维素(cap)、乙酸邻苯二甲酸纤维素、硝酸纤维素等纤维素酯类或它们的衍生物、聚偏氯乙烯、聚乙烯醇、聚乙烯乙烯醇、间规聚苯乙烯、聚碳酸酯、降冰片烯树脂、聚甲基戊烯、聚醚酮、聚酰亚胺、聚醚砜(pes)、聚苯硫醚、聚砜类、聚醚酰亚胺、聚醚酮酰亚胺、聚酰胺、氟树脂、尼龙、聚甲基丙烯酸甲酯、丙烯酸类聚合物或聚芳酯类、arton(商品名jsr社制造)或apelle(商品名三井化学社制造)之类的环烯烃类树脂等。在树脂膜的表面上,可以形成无机物、有机物的被膜或其两者的复合被膜,优选用以jisk7129-1992为基准的方法测定的、水蒸气透过度(25±0.5℃、相对湿度(90±2)%rh)为0.01g/m2·24h以下的阻隔性膜,进一步优选为用以jisk7126-1987为基准的方法测定的氧透过度为1×10-3ml/m2·24h·atm以下、水蒸气透过为1×10-5g/m2·24h以下的高阻隔性膜。作为形成阻隔膜的材料,只要是具有抑制带来水分或氧等元件所致劣化的物质的浸入的功能的材料即可,例如,可以使用氧化硅、二氧化硅、氮化硅等。另外,为了改良该膜的脆弱性,更优选具有由这些无机层和有机材料构成的层的叠层结构。对由无机层和有机层构成的层的叠层顺序没有特别限制,优选使两者交替地叠层多次。对阻隔膜的形成方法没有特别限定,可以使用例如真空蒸镀法、溅射法、反应性溅射法、分子线外延法、团簇离子束法、离子喷镀法、等离子体聚合法、大气压等离子体聚合法、等离子体cvd法、激光cvd法、热cvd法、涂敷法等,特别优选日本特开2004-68143号公报中所记载的利用大气压等离子体聚合法的方法。作为不透明的支撑基板,可列举例如:铝、不锈钢等金属板、膜或不透明树脂基板、陶瓷制成的基板等。本发明的有机el元件的发光在室温(25℃)下的外部导出量子效率优选为1%以上,更优选为5%以上。在此,外部导出量子效率(%)=有机el元件外部发光的光子数/流入有机el元件的电子数×100。另外,可以组合使用彩色滤光片等色相改良滤光片等,也可以组合使用颜色转换滤光片,所述颜色转换滤光片使用荧光体将来自有机el元件的发光色转变为多色。[密封]作为本发明的有机el元件的密封所使用的密封方法,可以列举例如将密封部件与电极、支撑基板用粘接剂粘接的方法。作为密封部件,只要以覆盖有机el元件的显示区域的方式配置即可,可以为凹板状,也可以为平板状。另外,透明性、电绝缘性没有特别限定。具体而言,可列举玻璃板、聚合物板/膜、金属板/膜等。作为玻璃板,特别可以列举钠石灰玻璃、含钡锶的玻璃、铅玻璃、铝硅酸玻璃、硼硅酸玻璃、钡硼硅酸玻璃、石英等。另外,作为聚合物板,可以列举聚碳酸酯、丙烯酸类聚合物、聚对苯二甲酸乙二醇酯、聚醚硫醚、聚砜等。作为金属板,可列举包含选自不锈钢、铁、铜、铝、镁、镍、锌、铬、钛、钼、硅、锗及钽中的1种以上的金属或合金的金属板。在本发明中,从可以将有机el元件薄膜化方面考虑,可以优选使用聚合物膜、金属膜。另外,聚合物膜优选为用以jisk7126-1987为基准的方法测定的氧透过度为1×10-3ml/m2·24h以下、用以jisk7129-1992为基准的方法测定的水蒸气透过度(25±0.5℃、相对湿度90±2%)为1×10-3g/m2·24h以下的膜。为了将密封部件加工成凹状,使用喷砂器加工、化学蚀刻加工等。作为粘接剂,具体而言,可以列举丙烯酸系低聚物、甲基丙烯酸系低聚物等具有反应性乙烯基的光固化及热固化型粘接剂、2-氰基丙烯酸酯等湿气固化型等粘接剂。另外,可以列举环氧系等热及化学固化型(二液混合)。另外,可以列举热熔型的聚酰胺、聚酯、聚烯烃。另外,可以列举阳离子固化型的紫外线固化型环氧树脂粘接剂。需要说明的是,有机el元件有时通过热处理而劣化,因此,优选可以在室温至80℃发生粘接固化的物质。另外,可以使干燥剂分散于所述粘接剂中。粘接剂向密封部分的涂布既可以使用市售的点胶涂敷机,也可以如丝网印刷那样进行印刷。另外,还可以优选在夹持有机el原件的构成层(例如发光层)并与支撑基板对向的一侧的电极的外侧被覆该电极和上述构成层,以与支撑基板连接的形态形成无机物、有机物的层而制成密封膜。该情况下,作为形成该膜的材料,只要是具有抑制水分或氧等元件所致劣化的物质的浸入的功能的材料即可,例如可以使用氧化硅、二氧化硅、氮化硅等。另外,为了改良该膜的脆弱性,优选具有由这些无机层和有机材料构成的层的叠层结构。对这些膜的形成方法没有特别限定,例如可以使用真空蒸镀法、溅射法、反应性溅射法、分子线外延法、团簇离子束法、离子喷镀法、等离子体聚合法、大气压等离子体聚合法、等离子体cvd法、激光cvd法、热cvd法、涂敷法等。优选在密封部件和有机el元件的显示区域的间隙中以气相及液相注入氮、氩等惰性气体或氟化烃、硅油那样的非活性液体。另外,也可以制成真空。另外,还可以在内部封入吸湿性化合物。作为吸湿性化合物,可列举例如:金属氧化物(例如氧化钠、氧化钾、氧化钙、氧化钡、氧化镁、氧化铝等)、硫酸盐(例如硫酸钠、硫酸钙、硫酸镁、硫酸钴等)、金属卤化物(例如氯化钙、氯化镁、氟化铯、氟化钽、溴化铈、溴化镁、碘化钡、碘化镁等)、过氯酸类(例如过氯酸钡、过氯酸镁等)等,在硫酸盐、金属卤化物及过氯酸类中,优选使用无水盐。[保护膜、保护板]为了提高元件的机械强度,可以在夹持有机el元件的构成层并与支撑基板对向的一侧的所述密封膜或所述密封用膜的外侧设置保护膜或保护板。特别是在利用所述密封膜进行密封的情况下,其机械强度不一定高,因此,优选设置这种保护膜、保护板。作为其中可以使用的材料,可以使用与用于所述密封的材料同样的玻璃板、聚合物板/膜、金属板/膜等,从轻量且薄膜化方面考虑,优选使用聚合物膜。[光导出提高技术]通常讲,有机el元件在折射率比空气高(折射率在1.6~2.1左右的范围内)的层的内部发光,仅导出在发光层中产生的光中的15%~20%左右的光。这是因为,以临界角以上的角度θ入射于界面(透明基板和空气的界面)的光发生全反射而无法导出至元件外部,或者,光在透明电极或发光层与透明基板之间发生全反射,光在透明电极或发光层进行波传导,结果,光向元件侧面方向散逸。作为提高该光的导出效率的方法,可列举例如在透明基板表面形成凹凸、防止透明基板和空气界面上的全反射的方法(例如美国专利第4774435号说明书)、通过使基板上具有集光性而提高效率的方法(例如日本特开昭63-314795号公报)、在元件的侧面等上形成反射面的方法(例如日本特开平1-220394号公报)、在基板和发光体之间导入具有中间折射率的平坦层并形成防反射膜的方法(例如日本特开昭62-172691号公报)、在基板和发光体之间导入折射率比基板低的平坦层的方法(例如日本特开2001-202827号公报)、在基板、透明电极层或发光层的任意层之间(包含基板和外界之间)形成衍射格子的方法(日本特开平11-283751号公报)等。在本发明中,可以将这些方法与本发明的有机el元件组合使用,可以优选使用在基板和发光体之间导入折射率比基板低的平坦层的方法、或在(包含)基板、透明电极层或发光层的任意层间(在内的基板与外界之间)形成衍射格子的方法。本发明通过组合这些方法,可以进一步得到高亮度或耐久性优异的元件。在透明电极和透明基板之间以比光的波长长的厚度形成低折射率的介质时,就从透明电极中出来的光而言,介质的折射率越低,导出至外部的效率越高。作为低折射率层,可列举例如:气凝胶、多孔质二氧化硅、氟化镁、含氟聚合物等。透明基板的折射率一般而言为1.5~1.7左右的范围内,因此,低折射率层的折射率优选为大约1.5以下。另外,进一步优选为1.35以下。另外,低折射率介质的厚度优选为介质中的波长的2倍以上。这是因为,低折射率介质的厚度为光的波长的程度、且为瞬逝渗出的电磁波进入于基板内的膜厚时,低折射率层的效果减弱。在引起全反射的界面或任一种介质中导入衍射格子的方法具有光导出效率的提高效果高的特征。该方法中,衍射格子通过1次衍射或2次衍射这样的所谓布拉格衍射,利用可以将光的方向变为与折射不同的特定的方向的性质,并通过在任意层间、或介质中(透明基板内或透明电极内)导入衍射格子,从而使从发光层中产生的光中由于层间全反射等而无法导出到外面的光发生衍射,将光导出至外部。导入的衍射格子优选具有二维的周期折射率。因为在发光层中发出的光在所有的方向随机地产生,因此,仅在某个方向上具有周期的折射率分布的通常的一维衍射格子中,仅有那些在特定方向上传导的光发生衍射,光的导出效率不会显著提高。但是,通过将折射率分布制成二维分布,在所有方向传导的光都发生衍射,光的导出效率提高。作为导入衍射格子的位置,可以为任意层间、或介质中(透明基板内或透明电极内),但优选产生光的场所即有机发光层的附近。此时,衍射格子的周期优选介质中的光波长的约1/2~3倍左右的范围内。衍射格子的排列优选重复正方形的格子状、三角形的格子状、蜂窝格子状等二维地排列。[集光片]本发明的有机el元件通过进行加工而在支撑基板(基板)的光导出侧设置例如微透镜阵列状的结构、或与所谓集光片组合,针对特定方向例如元件发光面进行正面方向集光,由此可以提高特定方向上的亮度。作为微透镜阵列的实例,在基板的光导出侧对一边为30μm、其顶角为90度的四棱锥进行二维排列。一边优选为10~100μm的范围内。比该范围小时,产生衍射效果而发生着色,过大时,厚度变厚,不优选。作为集光片,可以使用例如在液晶显示装置的led背光中被实用化的集光片。作为这种片材,例如可以使用住友3m社制造的亮度提高膜(bef)等。作为棱镜片的形状,例如既可以是在基材中形成了顶角90度、间距50μm的△状条纹的棱镜片,也可以是顶角带有圆润的形状、使间距随机地变化的形状、或其它形状。另外,为了控制来自有机el元件的光放射角,可以将光漫射板、膜与集光片组合使用。例如,可以使用(株)kimoto制造的扩散膜(lightup)等。[用途]本发明的有机el元件可以作为电子设备、例如显示装置、显示器、各种发光装置使用。作为发光装置,可列举例如:照明装置(家庭用照明、车内照明)、钟表或液晶用背光、广告牌、信号机、光存储介质的光源、电子照片复印机的光源、光通信处理机的光源、光传感器的光源等,但并不限定于此,特别可以有效地用于液晶显示装置的背光、作为照明用光源的用途。本发明的在有机el元件中,可根据需要在成膜时通过金属掩模或喷墨印刷法等实施图案化。图案化的情况下,可以仅将电极图案化,也可以将电极和发光层图案化,也可以将元件全层图案化,在元件的制作中,可以使用现有公知的方法。<显示装置>具备本发明的有机el元件的显示装置可以为单色,也可以为多色,在此,对多色显示装置进行说明。多色显示装置的情况下,仅在形成发光层时设置阴影掩模,可以在一面通过蒸镀法、流延法、旋涂法、喷墨法或印刷法等而形成膜。仅发光层进行图案化的情况下,并不限定于该方法,优选为蒸镀法、喷墨法、旋涂法及印刷法。显示装置中具备的有机el元件的构成根据需要从上述的有机el元件的构成例中选择。另外,有机el元件的制造方法如上述本发明有机el元件的制造中的一个实施方式所示。在对这样得到的多色显示装置施加直流电压的情况下,将阳极设为+的极性、将阴极设为-的极性并施加2~40v左右的电压时,可以观测发光。另外,即使以相反的极性施加电压,电流也不流动而完全不产生发光。在进一步施加交流电压的情况下,仅在成为阳极为+、阴极为-的状态时发光。需要说明的是,施加的交流的波形可以为任意的。多色显示装置可以作为显示设备、显示器或各种发光光源使用。在显示设备或显示器中,可以通过使用蓝、红及绿发光的3种有机el元件而进行全彩色的显示。作为显示设备或显示器,可列举电视机、笔记本电脑、移动设备、av设备、文字广播显示及汽车内的信息显示等。特别是可以作为再生静止图像或动态图像的显示装置使用,作为动画再生用显示装置使用时的驱动方式可以是简单矩阵(无源矩阵)方式,也可以是有源矩阵方式。作为发光装置,可列举家庭用照明、车内照明、钟表或液晶用背光、广告牌、信号机、光存储介质的光源、电子照片复印机的光源、光通信处理机的光源、光传感器的光源等,但本发明并不限定于这些。以下,基于附图对具有本发明的有机el元件的显示装置的一例进行说明。图7是表示由有机el元件构成的显示装置的一例的示意图。通过有机el元件的发光而进行图像信息的显示,例如是移动电话等的显示器的示意图。显示器10具有:具有多个像素的显示部a、基于图像信息而进行显示部a的图像扫描的控制部b、将显示部a和控制部b进行电连接的配线部c等。控制部b经由显示部a和配线部c而进行电连接,分别对多个像素基于来自外部的图像信息传输扫描信号和图像数据信号,每条扫描线上的像素依据扫描信号并按照图像数据信号依次发光并进行图像扫描而在显示部a显示图像信息。图8是有源矩阵方式的显示装置的示意图。显示部a在基板上具有含有多条扫描线15及数据线16的配线部c和多个像素13等。对显示部a的主要的部件的说明如下。在图8中,示出像素13发出的光向白箭头方向(下方向)导出的情况。配线部的扫描线15及多条数据线16分别由导电材料构成,扫描线15和数据线16以格子状垂直,在垂直的交点位置上与像素13连接(详细情况未图示)。像素13从扫描线15施加扫描信号时,由数据线16接收图像数据信号,根据接收的图像数据进行发光。通过将发光的颜色为红区域的像素、绿区域的像素、蓝区域的像素适当在同一基板上并置,可以进行全彩色显示。下面,对像素的发光工艺进行说明。图9是表示像素的电路的概略图。像素具备:有机el元件20、开关晶体管21、驱动晶体管22、电容器23等。在多个像素中使用红色、绿色及蓝色发光的有机el元件作为有机el元件20,可以通过将它们并列设置在同一基板上而进行全彩色显示。图9中,从控制部b经由数据线16对开关晶体管21的漏极施加图像数据信号。而且,从控制部b经由扫描线15对开关晶体管21的门极施加扫描信号时,开关晶体管21的驱动接通,施加于漏极的图像数据信号传递到电容器23和驱动晶体管22的门极。通过图像数据信号的传递,电容器23根据图像数据信号的电位而进行充电,同时驱动晶体管22的驱动接通。驱动晶体管22的漏极连接于电源线17,源极连接于有机el元件20的电极,根据施加于门极的图像数据信号的电位从电源线17向有机el元件20供给电流。通过控制部b的依次扫描而扫描信号移至下面的扫描线15时,开关晶体管21的驱动断开。但是,即使开关晶体管21的驱动断开,电容器23也保持被充电的图像数据信号的电位,因此,驱动晶体管22的驱动保持接通状态,继续有机el元件20的发光直至进行下面的扫描信号的施加。通过依次扫描施加下一扫描信号时,根据与扫描信号同期的下一图像数据信号的电位驱动驱动晶体管22而使有机el元件20发光。即,有机el元件20的发光相对于多个像素各自的有机el元件20设置作为主动元件的开关晶体管21和驱动晶体管22,多个像素13各自的有机el元件20发光。将这种发光方法称为有源矩阵方式。在此,有机el元件20的发光既可以为基于具有多梯度电位的多值图像数据信号的多梯度发光,也可以为基于2进制图像数据信号所规定的发光量的接通、断开。另外,电容器23的电位的保持既可以继续保持至下一扫描信号的施加,也可以在施加下一扫描信号之前进行放电。在本发明中,不限于上述的有源矩阵方式,可以为仅在扫描扫描信号时根据数据信号使有机el元件发光的无源矩阵方式的发光驱动。图10是无源矩阵方式的显示装置的示意图。图10中,多条扫描线15和多条数据线16夹持像素13而对向设置成格子状。通过依次扫描施加扫描线15的扫描信号时,与所施加的扫描线15连接的像素13根据图像数据信号进行发光。在无源矩阵方式中,在像素13中没有主动元件,可以实现制造成本的降低。通过使用本发明的有机el元件,可得到提高了发光效率的显示装置。<照明装置>本发明的有机el元件也可以用于照明装置。本发明的有机el元件可以用作具有共振器结构的有机el元件。作为具有这种共振器结构的有机el元件的使用目的,可列举光存储介质的光源、电子照片复印机的光源、光通信处理机的光源、光传感器的光源等,但并不限定于这些。另外,可以通过进行激光震荡而用于上述用途。另外,本发明的有机el元件既可以作为照明用途或曝光光源这样的一种灯使用,也可以作为投影图像这样类型的投影装置、或直接观看静止图像或动态图像这种类型的显示装置(显示器)使用。作为动画再生用显示装置使用时的驱动方式可以是无源矩阵方式,也可以是有源矩阵方式。或者,通过使用2种以上的具有不同的发光色的本发明的有机el元件,可以制作全彩色显示装置。另外,本发明所使用的π共轭系化合物可以适用于具备实质上产生白色发光的有机el元件的照明装置。例如,使用多个发光材料的情况下,可以通过使多个发光色同时发光并进行混色而得到白色发光。作为多个发光色的组合,既可以含有红色、绿色及蓝色的3原色的三个发光最大波长,也可以含有利用了蓝色和黄色、蓝绿和橙色等补色的关系的两个发光最大波长。另外,就本发明的有机el元件的形成方法而言,只要仅在发光层、空穴输送层或电子输送层等形成时设置掩模,利用掩模分涂等简单地配置即可。由于其它层为通用,因此,不需要掩模等图案化,可以在一面通过蒸镀法、流延法、旋涂法、喷墨法及印刷法等例如形成电极膜,生产率也提高。根据该方法,与以阵列状并列配置有多个色的发光元件的白色有机el装置不同,元件自身为白色发光。[本发明的照明装置的一个实施方式]对具备本发明的有机el元件的本发明的照明装置的一个实施方式进行说明。将本发明的有机el元件的非发光面用玻璃盒覆盖,将厚度300μm的玻璃基板用作密封用基板,在周围作为密封材料适用环氧类光固化型粘接剂(东亚合成社制luxtracklc0629b),将其重叠于阴极上并与透明支撑基板密合,从玻璃基板侧照射uv光并使其固化、密封,可以形成图11及图12所示的照明装置。图11表示照明装置的概略图,本发明的有机el元件(照明装置内的有机el元件101)用玻璃盖板102覆盖(需要说明的是,玻璃盖板中的密封操作不会使照明装置内的有机el元件101与大气接触而是在氮气氛下的手套箱(纯度99.999%以上的高纯度氮气的气氛下)中进行。)。图12表示照明装置的剖面图,105表示阴极,106表示有机el元件的构成层(例如发光层),107表示带透明电极的玻璃基板。需要说明的是,在玻璃盖板102内填充氮气108,设置有捕水剂109。通过使用本发明的有机el元件,可得到提高了发光效率的照明装置。<发光材料>本发明的π共轭类化合物可以通过电场激发等放射荧光。因此,可以作为各种发光材料使用。在发光材料中,除π共轭类化合物以外,可以根据需要含有其它成分。另外,发光材料可以以粉体状使用,也可以加工成所期望的形状而使用。发光材料可以适用于例如用于形成后述的发光性薄膜的材料、或荧光涂料、生物成像荧光色素。另外,π共轭类化合物的δest的绝对值为0.50ev以下时,有时π共轭类化合物放射延迟荧光,这种化合物可以适用于生物成像荧光色素。另外已知有延迟荧光通过氧而被消光,因此,也可以适用于氧传感材料。<发光性薄膜>本发明的发光性薄膜的特征在于,含有上述的本发明的π共轭类化合物,可以与构成所述有机el元件的各层的形成方法同样地制作。本发明的发光性薄膜可以与构成所述有机el元件的各层的形成方法同样地制作。本发明的发光性薄膜的形成方法没有特别限制,可以使用利用现有公知的例如真空蒸镀法、湿式法(湿工艺也称为)等的形成方法。作为湿式法,有旋涂法、流延法、喷墨法、印刷法、点胶涂敷法、刮板涂布法、辊涂法、喷涂法、流帘涂布法、lb法(langmuir-blodgett法)等,但从容易得到均质的薄膜、且高生产率方面考虑,优选点胶涂敷法、辊涂法、喷墨法、喷涂法等辊对辊方式适性高的方法。作为将本发明所使用的发光材料进行溶解或分散的液体介质,例如可以使用甲基乙基酮、环己酮等酮类、醋酸乙酯等脂肪酸酯类、二氯苯等卤化烃类、甲苯、二甲苯、均三甲苯、环己基苯等芳香族烃类、环己烷、十氢化萘、十二烷等脂肪族烃类、dmf、dmso等有机溶剂。另外,作为分散方法,可以通过超声波、高剪切力分散或介质分散等分散方法而进行分散。另外,每层可以使用不同的成膜法。在成膜中采用蒸镀法的情况下,其蒸镀条件因使用的化合物种类等不同而不同,但一般而言优选将舟加热温度在50~450℃的范围内、将真空度在1×10-6~1×10-2pa的范围内、蒸镀速度0.01~50nm/秒的范围内、基板温度-50~300℃的范围内、层厚0.1nm~5μm的范围内、优选5~200nm的范围内适当选择。另外,在成膜中采用旋涂法的情况下,优选在100~1000rpm的范围内、10~120秒的范围内、在干燥非活性气体气氛下进行旋涂。实施例以下,通过实施例具体地说明本发明,但本发明并不受实施例限定。需要说明的是,实施例中使用“%”的表示,但只要没有特殊说明,则表示“质量%”。以下示出实施例及比较例中使用的化合物。[化学式118][化学式119][化学式120][化学式121][化学式122][化学式123][化学式124][化学式125][化学式126][化学式127][化学式128][化学式129][化学式130][化学式131][化学式132][化学式133][化学式134][化学式135](化合物t-83的合成)使用国际公开第2010/113755号、organicletters,2002,4,1783-1785.、angew.chem.int.ed.2010,49,2014-2017.中所记载的方法,合成化合物t-83。具体而言,使1-溴-3,4,5-三氟苯和嘧啶衍生物进行交叉偶联反应,得到1-嘧啶基-3,4,5-三氟苯中间体。通过使1-嘧啶基-3,4,5-三氟苯中间体和溴嘧啶在钌催化剂存在下反应,得到1,2,3-三联嘧啶基-4,5,6-三氟苯中间体。通过使碱性条件下的咔唑和1,2,3-三联嘧啶基-4,5,6-三氟苯中间体反应,得到化合物t-83的粗纯化物。其后,进行色谱法、重结晶、升华纯化,得到化合物t-83的高纯度品。(其它的化合物的合成)与上述同样地操作,合成化合物t-2、t-3、t-13、t-27、t-33、t-59、t-66、t-74、t-78、t-79、t-82、t-83、t-84、t-85、t-96、t-101、t-117、t-124、t-125、t-131~t-133、t-176、t-180、t-182~t-208、t-210~t-212、t-214、t-216、t-221、t-227、t-240、t-249、t-252、t-254、t-257、t-265、t-275、t-282、t-298、t-306、t-328、t-353、t-354、t-365、t-366、t-399、t-429、t-432、t-434、t-442、t-447、t-453、t-456~t-459、t-497、t-504、t-511~t-520、t-522、t-525~t-527、t-530、t-531、t-540~t-553、t-555、t-556、t-558、t-561~t-574、及t-576。用以下的方法计算并求出得到的化合物及比较化合物1及2的δest。(δest的计算)就利用化合物的分子轨道计算进行的结构最适化及电子密度分布的计算而言,作为计算方法,采用使用b3lyp作为泛函数、使用6-31g(d)作为基底函数的分子轨道计算用软件来算出。作为分子轨道计算用软件,使用美国gaussian公司制造的gaussian09(revisionc.01,m.j.frisch,etal,gaussian,inc.,2010.)。由使用b3lyp作为该泛函数、使用6-31g(d)作为基底函数的结构最适化计算,进一步通过时间依赖密度泛函数法(time-dependentdft)实施激发状态计算来求出s1、t1的能级(分别为e(s1)、e(t1)),求出δest=|e(s1)-e(t1)|。[实施例1](有机el元件1-1的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行图案化之后,在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗之后,将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用坩埚填充最适于制作各元件的量的各层构成材料。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,对装有hat-cn(1,4,5,8,9,12-六氮杂三亚苯基六碳腈)的蒸镀用坩埚进行通电加热,以0.1nm/秒的蒸镀速度在ito透明电极上进行蒸镀,形成层厚10nm的空穴注入输送层。接着,以0.1nm/秒的蒸镀速度在所述空穴注入层上蒸镀α-npd(4,4′-双[n-(1-萘基)-n-苯基氨基]联苯),形成层厚40nm的空穴传输层。将主体化合物h-232、比较化合物1以分别成为94%、6%的体积%的方式以0.1nm/秒的蒸镀速度进行共蒸镀,形成层厚30nm的发光层。其后,将bcp(2,9-二甲基-4,7-二苯基-1,10-菲绕啉)以0.1nm/秒的蒸镀速度进行蒸镀,形成层厚30nm的电子传输层。另外,在以膜厚0.5nm形成氟化锂之后,蒸镀100nm的铝形成阴极。将上述元件的非发光面侧在纯度99.999%以上的高纯度氮气的气氛下、用罐状玻璃罩覆盖,设置电极导出布线,制作有机el元件1-1。(有机el元件1-2~1-141的制作)从比较化合物1开始,按照表1~4所示变更发光性化合物,除此之外,用与有机el元件1-1同样的方法制作有机el元件1-2~1-141。[表1][表2][表3][表4][实施例2](有机el元件2-1的制作)在100mm×100mm×1.1mm的玻璃基板上成膜100nm作为阳极的ito(铟锡氧化物)得到基板(nhtechnoglass公司制造的na45),对得到的基板进行图案化之后,在异丙醇中对带有该ito透明电极的透明支撑基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗。使用将聚(3,4-亚乙基二氧噻吩)-聚亚乙基磺酸酯(pedot/pss、bayer公司制造,baytronpal4083)用纯水稀释成70%而得到的溶液,在3000rpm、30秒的条件下利用旋涂法在该透明支撑基板上形成薄膜之后,在200℃下干燥1小时,设置层厚20nm的空穴注入层。将该透明支撑基板固定于市售的真空蒸镀装置的基板架上,在真空蒸镀装置内的各蒸镀用坩埚中填充最适于制作各元件的量的各层构成材料。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,以0.1nm/秒的蒸镀速度在所述空穴注入层上蒸镀α-npd,形成层厚40nm的空穴传输层。以0.1nm/秒的蒸镀速度对h-234、2,5,8,11-四叔丁基苝进行共蒸镀并使得分别成为97%、3%的体积%,形成层厚30nm的发光层。其后,以0.1nm/秒的蒸镀速度蒸镀tpbi(1,3,5-三(n-苯基苯并咪唑-2-基),形成层厚30nm的电子传输层。另外,在以膜厚1nm形成氟化钠之后,蒸镀100nm的铝形成阴极。将上述元件的非发光面侧在纯度99.999%以上的高纯度氮气的气氛下用罐状玻璃罩覆盖,设置电极导出配线,制作有机el元件2-1。(有机el元件2-2的制作)使用h-234作为主体化合物,使用2,5,8,11-四叔丁基苝作为发光性化合物,使用比较化合物1作为第三成分,以各自的比率成为82%、3%、15%的体积%的方式形成发光层,除此之外,与有机el元件2-1的制作同样地操作、制作有机el元件2-2。(有机el元件2-3~2-40的制作)如表5及6所示,改变第三成分,除此之外,用与有机el元件2-2同样的方法制作有机el元件2-3~2-40。[表5][表6][实施例3](有机el元件3-1的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行了图案化之后,在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗之后,将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用的电阻加热舟中填充最适于制作各元件的量的各层构成材料。所述电阻加热舟使用钼制或钨制。减压至真空度1×10-4pa之后,对放入有hat-cn的电阻加热舟进行通电而加热,以0.1nm/秒的蒸镀速度在ito透明电极上进行蒸镀,形成层厚15nm的空穴注入层。接着,以0.1nm/秒的蒸镀速度蒸镀α-npd(4,4’-双[n-(1-萘基)-n-苯基氨基]联苯),形成层厚30nm的空穴传输层。接着,对放入作为比较主体化合物的比较化合物1和gd-1的电阻加热舟进行通电加热,分别以0.1nm/秒的蒸镀速度、0.010nm/秒的蒸镀速度在所述空穴传输层上进行共蒸镀,形成层厚40nm的发光层。接着,以0.1nm/秒的蒸镀速度蒸镀hb-1,形成层厚5nm的第一电子传输层。进一步在其上以0.1nm/秒的蒸镀速度蒸镀et-1,形成层厚45nm的第二电子传输层。其后,以0.5nm的厚度蒸镀氟化锂之后,蒸镀100nm的铝形成阴极,制作有机el元件3-1。(有机el元件3-2~3-43的制作)从比较化合物1开始,按照表7及8所示变更主体化合物,除此之外,用与有机el元件3-1同样的方法制作有机el元件3-2~3-43。[表7][表8](发光效率的测定)有机el元件驱动时各样品的发光效率通过进行下述测定并进行评价。(相对发光效率的测定)使上述制作的各有机el元件在室温(约25℃)下、在2.5ma/cm2的恒定电流条件下发光,使用分光辐射亮度计cs-2000(柯尼卡美能达株式会社制造)测定发光刚开始之后的发光亮度。表1~8中示出得到的发光亮度的相对值(实施例1中为相对于有机el元件1-1的发光亮度的相对值,实施例2中为相对于有机el元件2-1的发光亮度的相对值,实施例3中为相对于有机el元件3-1的发光亮度的相对值)。在元件1~8中,任一种情况下,与比较例化合物相比,本发明的化合物均显示高的发光效率。[实施例4](有机el元件4-1的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行了图案化之后,在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗之后,将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用电阻加热舟中填充最适于制作各元件的量的各层构成材料。所述电阻加热舟使用钼制或钨制。减压至真空度1×10-4pa之后,对放入有α-npd(4,4’-双[n-(1-萘基)-n-苯基氨基]联苯)的电阻加热舟进行通电加热,以0.1nm/秒的蒸镀速度在ito透明电极上进行蒸镀,形成层厚30nm的空穴注入层。接着,以0.1nm/秒的蒸镀速度蒸镀tcta(三(4-咔唑基-9-基苯基)胺),形成层厚20nm的第一空穴传输层。另外,以0.1nm/秒的蒸镀速度蒸镀h-233,形成层厚10nm的第二空穴传输层。接着,对放入有作为比较主体化合物的比较化合物1和tbpe(2,5,8,11-四叔丁基苝)的电阻加热舟进行通电加热,分别以0.1nm/秒的蒸镀速度、0.010nm/秒蒸镀速度在所述空穴传输层上进行共蒸镀,形成层厚20nm的发光层。接着,以0.1nm/秒的蒸镀速度蒸镀h-232,形成层厚10nm的第一电子传输层。进一步在其上以0.1nm/秒的蒸镀速度蒸镀tbpi(1,3,5-三(1-苯基-1h-苯并咪唑-2-基)苯),形成层厚30m的第二电子传输层。其后,以0.5nm的厚度蒸镀氟化锂之后,蒸镀100nm的铝形成阴极,制作有机el元件4-1。(有机el元件4-2~4-49的制作)从比较化合物1开始,按照表9及10所示变更主体化合物,除此之外,用与有机el元件4-1同样的方法制作有机el元件4-2~4-49。(相对发光效率的测定)使上述制作的各有机el元件在室温(约25℃)下、在2.5ma/cm2的恒定电流条件下发光,使用分光辐射亮度计cs-2000(柯尼卡美能达株式会社制造)测定发光刚开始之后的发光亮度。表6中示出得到的发光亮度的相对值(实施例4中为相对于有机el元件4-1的发光亮度的相对值)。[表9][表10]在有机el元件4下,任一种情况下,与比较例化合物相比,本发明的化合物均显示高的发光效率。[实施例5]测定实施例5的元件1-19以初期亮度300cd/m2点亮时的亮度半衰期。(有机el元件5-1~5-47的制作)从实施例1的元件1-19开始,按照表11及表12所示变更主体化合物及发光性化合物,除此之外,用与有机el元件1-19同样的方法制作有机el元件5-1~5-47。[表11][表12]在有机el元件5中,在主体化合物为上述的通式(i)所表示的化合物的情况下,具有特别长的相对亮度半衰期。另外,在发光性化合物中,在供电子基团或吸电子基团3个以上连续地配置成邻位的情况下,具有长的相对亮度半衰期。[实施例6](共蒸镀膜6-1的制作)在异丙醇中对50mm×50mm、厚度0.7mm的石英基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗之后,将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用坩埚中填充1,3-双(n-咔唑基)苯(mcp)及发光性化合物t-124。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,以0.1nm/秒的蒸镀速度对1,3-双(n-咔唑基)苯(mcp)、发光性化合物t-124进行共蒸镀,并使得分别成为94%、6%的体积%,形成层厚40nm的共蒸镀膜。(共蒸镀膜6-2~4的制作)从t-124开始按照表13所示变更发光性化合物,除此之外,用与共蒸镀膜6-1同样的方法制作蒸镀膜6-2~6-4。[表13]共蒸镀膜发光材料6-1t-1246-2t-1806-3t-2116-4t-527(共蒸镀膜6-1~6-4的延迟荧光的测定)在氮气氛下对共蒸镀膜6-1~6-4照射355nm的激发光,测定它们在最大发光波长下的荧光减衰。关于各共蒸镀膜6-1~6-4,将表示时间和光子数的关系的曲线图示于图13~16。如图13~16所示,本发明的π共有化合物(t-124、t-180、t-211、及t-527)均确认到2种以上荧光的减衰速度不同的成分,确认了放射延迟荧光。以下,示出使用有特定的π共轭类化合物的实施例及比较例。以下示出以下的实施例及比较例中使用的化合物。<通式(201)所表示的π共轭类化合物>[化学式136][化学式137]<比较化合物>[化学式138]<其它的化合物>[化学式139]用以下的方法计算并求出上述化合物1~11及比较化合物1~3的δest。将其结果示于表14。(δest的计算)在利用化合物的分子轨道计算进行的结构最适化及电子密度分布的计算中,作为计算方法,采用使用b3lyp作为泛函数、使用6-31g(d)作为基底函数的分子轨道计算用软件来计算。作为分子轨道计算用软件,使用美国gaussian公司制造的gaussian09(revisionc.01,m.j.frisch,etal,gaussian,inc.,2010.)。由使用b3lyp作为该泛函数、使用6-31g(d)作为基底函数的结构最适化计算,进一步利用时间依赖密度泛函数法(time-dependentdft)进行激发状态计算而求出s1、t1的能级(分别为e(s1)、e(t1)),并按照δest=|e(s1)-e(t1)|求出δest。[表14]δest(ev)化合物10.18化合物20.16化合物30.19化合物40.09化合物50.14化合物70.05化合物60.10化合物80.16化合物90.36化合物100.29化合物110.13比较化合物10.01比较化合物20.34比较化合物30.12可以确认:通式(201)所表示的π共轭类化合物即化合物1~11的δest与比较化合物1(dmac-dps)、比较化合物2(2czpn)及比较化合物3(4czipn)的δest相比非常低,就显现tadf性而言,是足够小的值。[实施例7](有机el元件7-1的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行了图案化。在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗。使用将聚(3,4-亚乙基二氧噻吩)-聚亚乙基磺酸酯(pedot/pss、bayer公司制造、baytronpai4083)用纯水稀释成70%而成的溶液,在3000rpm、30秒的条件下利用旋涂法在该透明支撑基板上形成薄膜之后,在200℃下干燥1小时,设置膜厚20nm的空穴注入层。将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的蒸镀用坩埚中填充最适于制作各元件的量的各层构成材料。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,对放入有α-npd的蒸镀用坩埚中进行通电加热,以0.1nm/秒的蒸镀速度在ito透明电极上进行蒸镀,形成层厚30nm的空穴传输层。接着,以0.1nm/秒的蒸镀速度蒸镀将作为发光性化合物的dmac-dps(比较化合物1),形成层厚30nm的发光层。其后,以0.1nm/秒的蒸镀速度蒸镀ppt,形成层厚10nm的空穴阻挡层。进一步在其上以0.1nm/秒的蒸镀速度蒸镀化合物alq3,形成层厚30nm的电子传输层。另外,以0.5nm的膜厚蒸镀氟化锂而形成电子注入层,而后,蒸镀100nm的铝形成阴极。将上述元件的非发光面侧在纯度99.999%以上的高纯度氮气的气氛下、用罐状玻璃罩覆盖,设置电极导出配线,制作有机el元件7-1。(有机el元件7-2~7-9的制作)将发光性化合物变更为表2所示的化合物,除此之外,用与有机el元件7-1同样的方法制作有机el元件7-2~7-9。用以下的方法测定上述制作的各有机el元件的外部量子收率(eqe)及半衰期(连续驱动稳定性)。将评价结果示于表2。(外部量子收率(eqe)的评价)在室温(约25℃)下、在2.5ma/cm2的恒定电流条件下使上述制作的各有机el元件发光,使用分光辐射亮度计cs-2000(柯尼卡美能达株式会社制造)测定发光刚开始之后的发光亮度。将有机el元件1-1的亮度设为100,求出各有机el元件的亮度的相对值。(半衰期(连续驱动稳定性)的评价)以初期亮度1000cd/m2连续驱动各样品,同时使用上述分光辐射亮度计cs-2000测定亮度,求出测定的亮度减半的时间(lt50)。将有机el元件7-1的lt50设为100,求出各有机el元件的lt50的相对值,将其设为连续驱动稳定性的指标。将评价结果示于表15。表15中,表示eqe的数值越大,外部量子收率越高,表示半衰期的数值越大,连续驱动稳定性越优异(为长寿命)。[表15]如表15所示,使用了通式(201)所表示的π共轭类化合物的有机el元件7-4~7-9与使用了比较化合物1的有机el元件7-1相比,显示出具有不逊色的eqe,并且半衰期得以大幅度改善。另外,使用了通式(201)所表示的π共轭类化合物的有机el元件7-4~7-9与使用了比较化合物2及3的有机el元件7-2及7-3相比,显示出eqe和半衰期(特别半衰期)得以大幅度改善。[实施例8](有机el元件8-1的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行了图案化。在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗。将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用坩埚中填充最适于制作各元件的量的各层构成材料。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,对放入了dmac-dps(比较化合物1)作为发光性化合物的蒸镀用坩埚进行通电而加热,以0.1nm/秒的蒸镀速度在ito透明电极上进行蒸镀,形成层厚120nm的发光层。在得到的发光层上蒸镀100nm的铝形成阴极。将上述元件的非发光面侧在纯度99.999%以上的高纯度氮气的气氛下用罐状玻璃罩覆盖,设置电极导出配线,制作有机el元件8-1。(有机el元件8-2~8-9的制作)将发光性化合物变更为表16所示的化合物,除此之外,用与有机el元件8-1同样的方法制作有机el元件8-2~8-9。用与上述同样的方法测定上述制作的各有机el元件的外部量子收率(eqe)及半衰期(连续驱动稳定性)。而且,求出将有机el元件8-1的eqe及半衰期设为100时的各有机el元件的相对值。将评价结果示于表16。[表16]如表16所示,使用了通式(201)所表示的π共轭类化合物的有机el元件8-4~8-9与使用了比较化合物的有机el元件8-1~8-3相比,显示出可以大幅度改善eqe和半衰期,特别是半衰期。[实施例9](有机el元件9-1的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行了图案化。在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗。将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用坩埚中填充最适于制作各元件的量的各层构成材料。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,对放入了要蒸镀的化合物的蒸镀用坩埚进行通电加热,以0.1nm/秒的蒸镀速度将发光层的主体化合物和客体化合物按照表4中记载的比例进行共蒸镀,形成层厚120nm的发光层。在得到的发光层上蒸镀100nm的铝形成阴极。将上述元件的非发光面侧在纯度99.999%以上的高纯度氮气的气氛下用罐状玻璃罩覆盖,设置电极导出配线,制作有机el元件9-1。(有机el元件9-2的制作)将发光性化合物变更为表17所示的化合物,除此之外,用与有机el元件9-1同样的方法制作有机el元件9-2。用与上述同样的方法测定上述制作的各有机el元件的半衰期(连续驱动稳定性)。而且,将有机el元件9-1的半衰期设为100,求出有机el元件9-2的半减期的相对值。将评价结果示于表4。[表17]如表17所示,组合了通式(201)所表示的π共轭类化合物和客体化合物的有机el元件9-2与没有组合有客体化合物的有机el元件9-1相比,显示出半衰期得到进一步改善。(有机el元件9-3的制作)在50mm×50mm、厚度0.7mm的玻璃基板上以150nm的厚度成膜作为阳极的ito(铟锡氧化物),进行图案化。在异丙醇中对带有该ito透明电极的透明基板进行超声波清洗,用干燥氮气进行干燥,进行5分钟uv臭氧清洗。将该透明基板固定于市售的真空蒸镀装置的基板架上。在真空蒸镀装置内的各蒸镀用坩埚中填充最适于制作各元件的量的各层构成材料。蒸镀用坩埚使用由钼制或钨制的电阻加热用材料制作的坩埚。减压至真空度1×10-4pa之后,对放入了要蒸镀的化合物的蒸镀用坩埚进行通电而加热,以0.1nm/秒的蒸镀速度将发光层的主体化合物和客体化合物按照表18中记载的比例进行共蒸镀,形成层厚110nm的发光层。在得到的发光层上蒸镀100nm的铝形成阴极。将上述元件的非发光面侧在纯度99.999%以上的高纯度氮气的气氛下用罐状玻璃罩覆盖,设置电极取出配线,制作有机el元件9-3。(有机el元件9-4~9-5的制作)将发光性化合物变更为表5所示的化合物,除此之外,用与有机el元件9-3同样的方法制作有机el元件9-4~9-5。用与上述同样的方法测定上述制作的各有机el元件的外部量子收率(eqe)。而且,将有机el元件3-3的eqe的值设为100,求出有机el元件9-4及5的eqe的相对值。将评价结果示于表18。[表18]如表18所示,发光层中的客体化合物的掺杂材浓度少于20体积%的有机el元件9-4~9-5与发光层中的客体化合物的掺杂材浓度超过20体积%的有机el元件9-3相比,显示出eqe进一步得以改善。本申请主张基于2015年5月8日提出申请的日本特愿2015-095804号、2015年10月15日提出申请的日本特愿2015-203876号、及2015年10月15日提出申请的日本特愿2015-203878号的优先权。这些申请说明书及附图中所记载的内容在全部本申请说明书中被引用。工业上的可利用性根据本发明,可以提供例如可以改良有机电致发光元件的发光效率的新型的π共轭类化合物。另外,可以提供使用了该π共轭类化合物的有机电致发光元件材料、发光性薄膜、发光材料、有机电致发光元件、以及具备该有机电致发光元件的显示装置及照明装置。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1