IL‑37变体的制作方法
相关申请的交叉引用
本申请要求澳大利亚临时申请2015902262和2016900703的优先权,这两篇申请的全部内容通过引用并入本文。
发明领域
本发明涉及多肽,其包括白介素-37(il-37)的变体,和相关治疗剂和组合物。本发明还涉及多肽和组合物在治疗炎性疾病或病症的方法中的用途。
发明背景
炎症在宿主防御和免疫介导的疾病的进展中起着基本作用。炎症反应响应于组织损伤(例如创伤、局部缺血和外来颗粒)和由包括化学介质(例如细胞因子和前列腺素)和炎性细胞(例如白细胞)在内的复杂级联事件的感染而启动。炎症反应的特征在于血流量增加、毛细血管通透性增加和吞噬细胞的流入。这些事件导致肿胀、发红、温暖(改变的热量模式),并在损伤部位形成脓液。
炎症反应中体液和细胞免疫要素之间的相互作用使得能够消除有害物质并开始修复受损组织。当这种相互作用被破坏时,炎症反应可能导致正常组织受到不受控制的炎症反应的相当大的损害,需要临床干预以阻止组织损伤和器官功能障碍。
白介素-37(il-37)是il-1细胞因子家族的一员,在先天性和获得性免疫中具有独特和广泛的抗炎作用。il-37通过促炎细胞因子如il-1β和tnf经它们的受体以及toll样受体配体的介导特异性地减少炎症,并且在由感染或其他非感染性攻击触发的炎症中具有广泛的保护作用。il-37既通过与il-18受体α和il-1r8(sigirr)的相互作用在细胞膜上启动信号传导,并且还通过与smad3的相互作用在细胞内启动信号传导。然而,目前还缺乏对il-37活性的结构、功能和调节之间关系的理解。
存在对改进的抗炎组合物和治疗方法的需求。
本说明书中的任何现有技术的参考不是承认或暗示该现有技术形成任何辖区的众所周知常识的一部分,或者该现有技术可合理预期被理解为、被认为是相关的和/或与本领域技术人员已知的其他现有技术组合。
发明概述
本发明提供单体抗炎多肽,其包含il-37单体的氨基酸序列,所述氨基酸序列具有阻止抗炎多肽形成同二聚体的突变或修饰。
本发明提供抗炎多肽,其包含il-37多肽的氨基酸序列,所述氨基酸序列具有降低所述抗炎多肽形成二聚体的能力的突变或修饰。
优选地,突变或修饰减少或防止多肽形成能够使il-37单体二聚化的二聚化界面。通常,突变或修饰位于肽的与形成il-37单体的二聚化界面的氨基酸序列具有相同氨基酸序列的区域中。突变或修饰可位于形成il-37单体的二聚化界面的β3或β4环中。
本发明提供了多肽,其包含il-37多肽或其片段的氨基酸序列,其中与具有seqidno:1的序列的多肽相比,所述多肽具有降低的形成二聚体的能力。
本发明提供了多肽,其包含与seqidno:1具有至少50%、60%、70%、75%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%的序列同一性的氨基酸序列,其中与具有seqidno:1的序列的多肽相比,所述多肽具有降低的形成二聚体的能力。
本发明提供了il-37多肽,其与具有seqidno:1的序列的多肽相比,具有降低的形成二聚体的能力。
本发明提供了多肽,其包含il-37多肽或其片段的氨基酸序列,其中与具有seqidno:1的序列的多肽相比,所述多肽具有降低的形成二聚体的能力,其中所述多肽具有位于等价于seqidno:1中的β3-β4环区域的位置中的至少一个残基的突变或修饰。优选地,β3-β4环区域由seqidno:1的残基83至91组成或者等价于其的残基组成。优选地,所述突变或修饰发生在k83、n84、y85、i86、r87和/或p88的位置或等价于其的位置的残基上。优选地,所述突变是用非保守残基进行的替换,更优选地,所述突变是用丙氨酸或具有相反电荷的氨基酸进行的替换。
本发明提供了多肽,其包含il-37多肽或其片段的氨基酸序列,其中与具有seqidno:1的序列的多肽相比,所述多肽具有降低的形成二聚体的能力,其中所述多肽具有在d73、k83和y85的位置或等价于其的位置的残基上的突变或修饰。优选地,所述突变是用非保守氨基酸残基进行的替换,更优选地,所述突变是用丙氨酸或具有相反电荷的氨基酸进行的替换。更优选地,突变是d73a、d73k、k83e、k83a和y85a中的任何一个或多个。
本发明提供了多肽,其包含il-37多肽或其片段的氨基酸序列,其中与具有seqidno:1的序列的多肽相比,所述多肽具有降低的形成二聚体的能力,其中所述多肽具有v71、v80和i78的位置或等价于其的位置的残基上的突变或修饰。优选地,所述突变是用非保守残基进行的替换,更优选地,所述突变是用丙氨酸或具有相反电荷的氨基酸进行的替换。
本发明提供了il-37多肽,其与具有seqidno:1的序列的多肽相比具有降低的形成二聚体的能力,其中所述多肽具有与seqidno:1中的β3-β4环区域等价的区域的修饰。修饰包括缺失环、缩短环、延长环、突变环的残基和/或化学修饰环残基。
本发明提供了一种单体多肽,其包含与seqidno:1具有至少50%、60%、70%、75%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%的序列同一性的氨基酸序列。
本发明提供了作为单体存在的分离的、重组的或合成的il-37多肽。
本发明提供了包含seqidno:1所示序列的旁系同源序列或直系同源序列的多肽,其中所述多肽作为单体存在或与具有seqidno:1序列的多肽相比具有降低的形成二聚体的能力。优选地,包含seqidno:1所示序列的旁系同源或直系同源序列的多肽在位于二聚化界面的残基中具有突变或修饰,所述二聚化界面等价于用于seqidno:1的本文所描述的二聚界面,例如表1中的残基。
本发明的多肽可以是分离的、纯化的、基本上纯化的、富集的、合成的或重组的。
如本文所用,降低的形成二聚体的能力可以是指降低的形成异二聚体和/或同二聚体的能力。
本发明还提供了多肽,其包含il-37多肽或其片段的氨基酸序列,基本由其组成或由其组成,其中所述氨基酸序列含有seqidno:1中二聚体界面的残基、或等价于其的位置的至少一个突变或修饰。与具有seqidno:1的氨基酸序列的多肽有关的二聚体界面包括表1中的残基。通常,所述多肽包含氨基酸序列,其包含在seqidno:1的73、83和85位或等价于其的位置残基的突变或修饰。更优选地,突变是d73a、d73k、k83e、k83a和y85a中的任一个或多个。
本发明提供了分离的、重组的或合成的il-37多肽,其不具有形成二聚体的能力或具有降低的形成二聚体的能力。在本文所述的本发明的这个或任何其他方面,多肽形成二聚体的能力可通过本文所述的任何方法来测定,包括尺寸排阻色谱和多角度光散射,在非变性条件下的分析凝胶电泳、分析离心、质谱或反相高效液相色谱(rp-hplc)。
二聚体形成的减少可以与参考多肽相比较。参考多肽通常是天然的、野生型的、未修饰的或未突变的il-37多肽。优选地,参考多肽具有seqidno:1的氨基酸序列。或者,如果本发明的多肽具有与seqidno:1旁系同源或直系同源的氨基酸序列,则参考肽是天然的、野生型的、未修饰的或未突变的旁系同源或直系同源氨基酸序列。
本发明提供了包含与seqidno:1具有至少50%、60%、70%、75%、80%、81%、82%、83%、84%、85%、86%、87%、88%、89%、90%、91%、92%、93%、94%、95%、96%、97%、98%或99%的序列同一性的氨基酸序列,其中在下述位置或等价于其的位置的氨基酸残基:
seqidno:1中的位置71不是val;
seqidno:1中的位置72不是leu;
seqidno:1中的位置73不是asp;
seqidno:1中的位置74不是ser;
seqidno:1中的位置78不是ile;
seqidno:1中的位置80不是val;
seqidno:1中的位置83不是lys;
seqidno:1中的位置84不是asn;
seqidno:1中的位置85不是tyr;
seqidno:1中的位置86不是ile;
seqidno:1中的位置87不是arg;
seqidno:1中的位置88不是pro;和/或
seqidno:1中的位置184不是asn。优选地,所述氨基酸残基是相对于在seqidno:1中的该位置出现的氨基酸的非保守置换。在一个实施方案中,位置85的氨基酸是丙氨酸,位置83的氨基酸是谷氨酸,位置73的氨基酸是丙氨酸和/或位置73的氨基酸是赖氨酸。在一个实施方案中,在seqidno:1中的位置71、72、73、74、78、80、83、84、85、86、87、88和/或184的位置上或在等价于其的位置上的氨基酸残基是缺失的。
关于本文所述的本发明的任何多肽,所述多肽可具有seqidno:1的1至20或1至45或等价于其的残基的n末端截短。
与具有seqidno:1的氨基酸序列,未修饰或未突变的il-37氨基酸序列或不表现出降低的形成二聚体的能力的il-37多肽的多肽相比,本文所述的本发明的任何多肽还可展现出更大的抗炎性质。抗炎性质可以通过本文描述的测定来确定,特别是在实施例中的,包括实施例1、3和4。在一个实施方案中,当在包括实施例1和3中所述的lps刺激的il-1β测定的本文所述的测定中测试时,本发明的多肽在浓度小于或约1ug/ml、100ng/ml、10ng/ml、100pg/ml、10pg/ml或1pg/ml时显示出抗炎特性。炎性损伤可以包括toll样受体(tlr)2、4、7、8和/或9的配体,如hklm、咪喹莫特、cpg-a或ssrna40。当在实施例1和3所述的lps刺激的il-1β测定中测试时,本发明的多肽可以在10pg/ml减少il-1β至少30%,在10ng/ml减少il-1β至少40%或在1pg/ml、10pg/ml、100pg/ml、10ng/ml或100ng/ml减少至少约50%。还可以观察到其它促炎性细胞因子如il-6的减少。当在本文所述的包括实施例1和3中所述的lps刺激的il-1β测定的测定中以10ng/ml和100ng/ml进行测试时,本发明的多肽在抗炎性质方面可以不显示统计学上的显著差异。本发明的多肽可以表现出与实施例中描述的突变的il-37相同或相似的抗炎性质。
本发明提供了用于治疗或预防炎性疾病或病症的药物组合物,其包含本发明的多肽和药学上可接受的稀释剂、赋形剂或载体。在一个实施方案中,组合物中存在的唯一活性成分是本发明的多肽。
本发明提供用于治疗或预防炎性疾病或病症的药物组合物,其包含作为活性成分的本发明的多肽和药学上可接受的稀释剂、赋形剂或载体。在一个实施方案中,组合物中存在的唯一活性成分是本发明的多肽。
本发明提供用于治疗或预防炎性疾病或病症的药物组合物,其包含作为主要成分的本发明的多肽和药学上可接受的稀释剂、赋形剂或载体。在一个实施方案中,组合物中存在的唯一活性成分是本发明的多肽。
本发明还提供了本发明的多肽,其用于治疗炎性疾病或病症。
本发明还提供了包含本发明多肽和药学上可接受的稀释剂、赋形剂或载体的药物组合物,其用于治疗炎性疾病或病症。
本发明还提供了抑制有此需要的受试者中的炎症的方法,所述方法包括向所述受试者施用治疗有效量的本发明的多肽或本发明的药物组合物,从而抑制炎症。
本发明提供了用于治疗或预防炎性疾病或病症的方法,所述方法包括向需要治疗或预防的受试者施用组合物的步骤,其中所述组合物包含本发明的多肽和药学上可接受的稀释剂、赋形剂或载体,基本上由其组成或由其组成。
在本文所述的本发明的任何方法或用途中,本发明的多肽可全身地或直接地施用于疾病部位。本发明的多肽可以配制用于口服给药。
本发明提供了治疗或预防有需要的受试者中的炎性疾病或病症的方法,所述方法包括向所述受试者施用治疗有效量的本发明的多肽或药物组合物,由此治疗或预防受试者中的炎性疾病或病症。
本发明还提供了缓解或改善有需要的受试者的炎性疾病或病症的症状的方法,所述方法包括向有需要的受试者施用治疗有效量的本发明的多肽或药物组合物,由此减轻或改善受试者中炎性疾病或病症的症状。
本发明还提供了治疗有效量的本发明的多肽或药物组合物在制备用于治疗或预防有需要的受试者中的炎性疾病或病症的药物中的用途。
本发明提供了用于治疗受试者中的炎性疾病或病症的方法,其包括以下步骤:
鉴定患有炎性疾病或病症的受试者;和
给有此需要的受试者施用治疗有效量的本发明的多肽或药物组合物,
由此治疗受试者中的炎性疾病或病症。
本发明提供了治疗炎性疾病或病症的方法,其包括以下步骤:
鉴定患有炎性疾病或病症的受试者;和
给有此需要的受试者施用治疗有效量的本发明的多肽或药物组合物,
由此治疗受试者中的炎性疾病或病症。
本发明还提供编码如本文所述的多肽的核酸分子。
本发明还提供了包含本文所述的核酸分子的载体。
本发明还提供了包含本文所述的载体或核酸分子的细胞。
本发明还提供了由其衍生的动物或组织,其包含本文所述的细胞。
如本文所用,除上下文另有要求外,术语“包含(comprise)”以及该术语的变化形式例如“包含(comprising)”、“包含(comprises)”和“包含(comprised)”不旨在排除其它添加物、组分、整数或步骤。术语“包括”也可以与“包含”互换使用,并且也不旨在排除其他添加物、组分、整数或步骤。
通过以下通过示例并参考附图给出的描述,本发明的其他方面以及前面段落中所述方面的进一步实施方案将变得显而易见。
附图简要说明
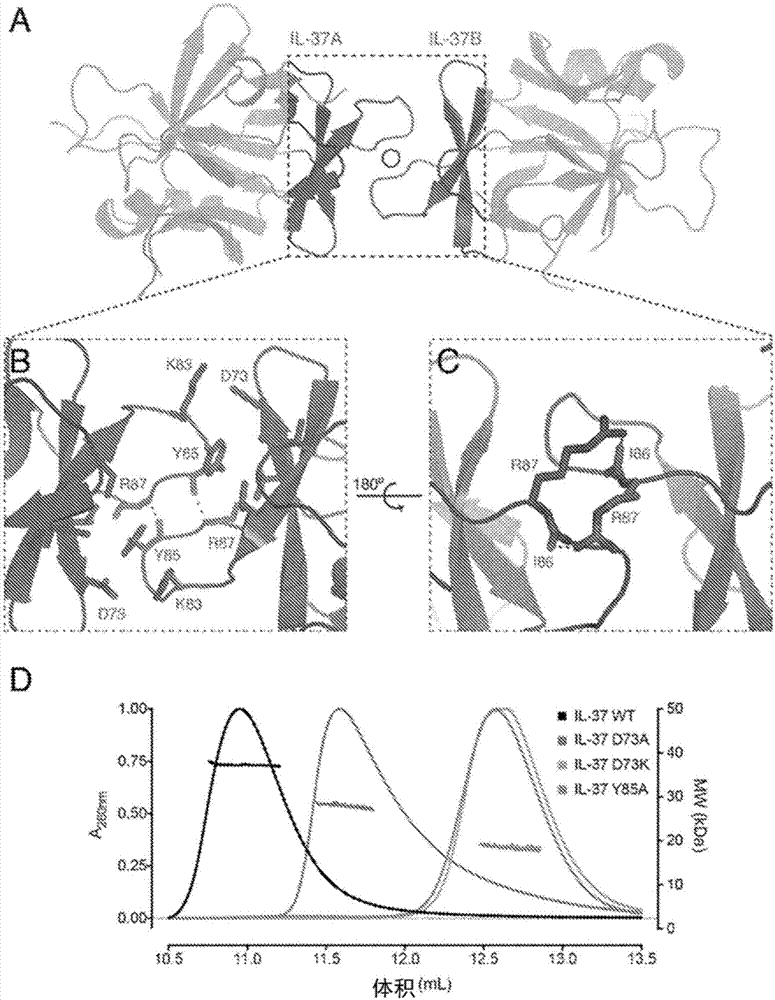
图1:il-37形成了一种新型的头对头对称同二聚体。(a)纯化的野生型il-37(1-218)和n末端截短变体il-37(21-218)和il-37(46-218)的sds-page。示出了一小部分sds稳定的il-37二聚体。(b)相对于单体il-18的il-37同二聚体的sec-mals分析。sec迹线对应于左侧y轴,且平均分子量的mals确定(右侧y轴)在折射率迹线下方以虚线显示。(c)从两个方向观察的il-37同二聚体的概况,相对于水平轴顺时针旋转90°。结构以动画(cartoon)格式显示,左边是il-37a,右边是il-37b(a和b用于区分同二聚体中的il-37分子)。
图2:il-37二聚体界面的结构和突变分析。(a)动画格式的il-37同二聚体,同二聚体的c2对称轴标记为圆形。(b)具有侧链的同二聚化界面,使关键相互作用显示为棒状而氢键显示为虚线。(c)il-37界面相对于(b)围绕水平旋转180°,关键侧链为棒状。(d)il-37二聚体界面突变体的sec-mals分析,sec迹线对应于左侧y轴,且平均分子量的mals确定(右侧y轴)在折射率迹线下方以虚线显示。
图3:il-37二聚体界面的secmals。野生型il-37和il-37界面突变体的sec-mals分析。通过280nm处的吸光度(左轴)监测sec,并且右轴指示来源于mals的平均分子量。*应当指出,位于二聚体和野生型峰之间的峰是通过柱时解离的二聚体的特征(参见woodbury等人,proteinscience2002),即在sec运行期间蛋白质在单体和二聚体之间不是处于平衡状态,而是随着浓度在柱上稀释时降低而不断解离。
图4:il-37同种型1(seqidno:1)。登录号np_055254。这是本文使用的il-37同种型的全长序列并且与本文中提及的编号相关。用于晶体试验的序列来自该序列的残基46-218。
图5:人pbmc中重组il-1β变体对lps刺激的il-1β的不同抑制作用。将从健康志愿者新鲜分离的pbmc与指定浓度的重组il-37b的四种变体(包括天然蛋白(野生型)和单体突变体(d73k)、在氨基酸21(21-218)或46(46-218)截短的两个n末端截短体)孵育。30分钟后,用lps(50pg/ml)或媒介物(无il-1β诱导,未显示)刺激培养物。加入lps后20小时收集上清液,并通过elisa检测il-1β。图显示与lps+媒介物(设为0)相比,由il-37b变体提供的il-1β改变百分比±sem;n=15个供体;*p<0.05,媒介物+lps相对于所有其他组;#p<0.05和##p<0.01,相同浓度的单体il-37相对于所有其他变体。
图6:通过转染到thp-1巨噬细胞中的il-37b变体减少il-1β。用指定的il-37b变体(全长,1-218)转染thp-1巨噬细胞,然后用pma分化细胞,随后用lps(250ng/ml)或媒介物刺激。24小时后收集上清液并通过elisa测定il-1β。上清液il-1β±sem;n=11-26;*p<0.05,对照相对于il-37b转染;#p<0.05,天然il-37b相对于单体il-37。与对照转染相比,转染il-37b变体的p<0.001。
图7:在10mg/kglps或媒介物(时间0h)之前60分钟,向c57bl/6野生型(wt)小鼠腹膜内注射重组il-37变体(40μg/kg)或媒介物。il-37tg小鼠还注射lps或媒介物以直接比较。总共n=94只小鼠,wt的每组4-24只,il-37tg的5只。在指定的时间点测量体温并绘制±sem;所有的媒介物小鼠一起显示。*,p<0.05和***,p<0.001,lps相对于所有其他条件;#,p<0.05;##,p<0.01和###,p<0.001,d73k或y85a变体相对于天然recil-37;ns-tg,相对于il-37tg不显著;ns-v,相对于媒介物不显著;*1-4,7小时统计:*1,*和ns-v;*2,*和ns-v和ns-tg;和*3,***和ns-tg;*4,*和ns-tg。recil-3721-218显示为灰色用于直接比较,这些组的统计分开计算。
图8:根据图7,19小时时wt小鼠中的血浆il-1β±sem。veh,媒介物;nat,天然recil-37;mono,d73k;数字表示变体的n末端截短。
图9:人pbmc中重组il-37变体对hklm刺激的il-1β或il-6的不同抑制作用。如所示,刺激新鲜分离的pbmc。使用hklm(热灭活的单核细胞增生李斯特菌),每毫升106个细胞,在recil-37变体后30分钟加入。黑色,单独的hklm;白色,hklm+天然recil-37;浅灰色,hklm+d73k;深灰色,hklm+y85a(所有变体46-218)。在加入hklm后20小时收集上清液,并通过elisa测量il-1β(a-c,g)和il-6(d-f,h)。(a-f)浓度(conc)1,10ng/ml;浓度2,100pg/ml;浓度3,10pg/ml。图显示个体供体fc(a,d),jm(b,e)和lm(c,f)培养物的上清液中的绝对细胞因子浓度±sem。(e-f)hklm+d73k浓度1的数据不可用。(g,h)由图a-f所示的原始数据计算il-1β(g)和il-6(h)的百分比改变,并描绘为±sem。recil-37的浓度以pg/ml表示。*,p<0.05;**,p<0.01;***p<0.001,仅hklm相对于hklm+recil-37;##p<0.01,相同浓度的y85a相对于天然recil-37。
图10:人pbmc中由重组il-37变体对咪喹莫特刺激的il-1β或il-6的不同抑制作用。如所示,刺激新鲜分离的pbmc。咪喹莫特以10μg/ml使用,并在recil-37b变体后30分钟加入。黑色,单独的咪喹莫特;白色,咪喹莫特+天然recil-37;浅灰色,咪喹莫特+d73k;深灰色,咪喹莫特+y85a(所有变体46-218)。加入咪喹莫特20h后收集上清液,并通过elisa测量il-1β(a-c,g)和il-6(d-f,h)。(a-f)浓度(conc)1,10ng/ml;浓度2,100pg/ml;浓度3,10pg/ml。图显示个体供体fc(a,d),jm(b,e)和lm(c,f)培养物的上清液中的绝对细胞因子浓度±sem。(e)咪喹莫特+天然浓度2的数据不可用。(g,h)由图a-f所示的原始数据计算il-1β(g)和il-6(h)的百分比改变,并描绘为±sem。recil37的浓度以pg/ml表示。*,p<0.05;**,p<0.01,单用咪喹莫特相对于咪喹莫特+recil37;#,p<0.05,相同浓度的y85a相对于天然recil-37。
图11:如所示,刺激新鲜分离的pbmc。cpg-a以3μm使用,并在recil-37b变体后30分钟加入。黑色,单独的cpg-a;白色,cpg-a+天然recil-37;浅灰色,cpg-a+d73k;深灰色,cpg-a+y85a(所有变体46-218)。在加入cpg-a后20小时收集上清液,并通过elisa测量il-6(a-c)。(a-c)浓度(con)1,10ng/ml;浓度2,100pg/ml;浓度3,10pg/ml。图显示个体供体fc(a),jm(b)和lm(c)的培养物的上清液中绝对细胞因子浓度±sem。cpg-a+d73k浓度3的数据不可用。(d)从图a-c所示的原始数据计算il-6的百分比改变,并描绘为±sem。recil37的浓度以pg/ml表示。*,p<0.05;**,p<0.01,单独的cpg-a相对于cpg-a+recil37。
图12:如所示,刺激新鲜分离的pbmc。ssrna40以0.5μg/ml使用,并在recil-37变体后30分钟加入。黑色,单独的ssrna40;白色,ssrna40+天然recil-37;浅灰色,ssrna40+d73k;深灰色,ssrna40+y85a(所有变体46-218)。加入ssrna40后20小时收集上清液,并通过elisa测定il-1β。(a,b)浓度(conc)1,10ng/ml;浓度2,100pg/ml;浓度3,10pg/ml。图显示个体供体jf(a)和ms(b)的培养物的上清液中绝对细胞因子浓度±sem。ssrna40+d73k浓度3(jf)和2(ms)的数据不可用。(c)从图a和b所示的原始数据计算il-1β的百分比改变,并描绘±sem。recil37的浓度以pg/ml表示。没有进行统计计算,因为只有两个可用的数据集。
实施方案的详述
应该理解的是,在本说明书中公开和限定的本发明延伸到从文本或附图中提到或明显可见的两个或更多个单独特征的所有替代组合。所有这些不同的组合构成本发明的各种替代方面。
现在将详细参考本发明的某些实施方案。尽管将结合实施方案描述本发明,但是应该理解的是,本发明并不将本发明限制于那些实施方案。相反,本发明旨在覆盖可以包括在由权利要求书限定的本发明的范围内的所有替代、修改和等价物。
本领域技术人员将认识到许多与本文所述相似或等价的方法和材料,其可用于实施本发明。本发明决不限于所描述的方法和材料。应该理解的是,在本说明书中公开和限定的本发明延伸到从文本或附图中提到或明显可见的两个或更多个单独特征的所有替代组合。所有这些不同的组合构成本发明的各种替代方面。
在此引用的所有专利和出版物均通过引用整体并入。
为了解释本说明书,以单数使用的术语也将包括复数,反之亦然。
导致本发明的本发明人的工作包括il-37的
本发明的多肽表现出显著增加的抗炎活性。本发明的多肽的优点在于在增加的浓度下野生型(即天然的、内源的)il-37观察到的功效损失不会发生。换句话说,本发明的多肽在高浓度和超过至少4log浓度范围内持续表现出抗炎作用。这是特别有利的,因为与野生型重组il-37相比,本发明多肽的治疗窗口相当宽。
il-37多肽是具有il-37的至少一种生物化学或生物物理活性的分子,例如其可以结合白介素18受体(il-18r1/il-1rrp)并且可以是白介素18受体的配体。它还可以结合白介素18结合蛋白(il-18bp),即白介素18(il-18)的抑制性结合蛋白,并且随后与il-18受体β链形成复合物,并且通过该复合物它可以抑制il-18活性。il-37的其他生物化学或生物物理活性包括与il-1r8(sigirr)的结合,阻断在人或鼠免疫细胞中由广谱炎症攻击(包括tlr配体,ifnγ,tnf和il-1β)触发的促炎细胞因子(而非抗炎细胞因子)的产生,抑制树突细胞的活化(减少cd86和mhcii的表面表达),触发细胞内激酶的特异性调控模式(包括阻断mtor、mapk和nf-κb通路),和诱导抗炎激酶如mer和pten(如(5)中所述)。
il-37也被称为白介素-37(fil1ζ;il-1ζ;il-1f7b(il-1h4、il-1h、il-1rp1);il-1x蛋白;il1f7(标准产物il-1f7b);白介素1家族成员7;白介素1,ζ;白介素-1同系物4;白介素-1超家族z;白介素-1相关蛋白和白介素-23)。人il-37具有5种同种型,a、b、c、d和e,当涉及本文中的il-37时,除非另有明确说明,否则所有这些均包括在内。包含至少一个与表1中的二聚体界面残基等价的残基的人il-37多肽的任何同种型或直系同源物也包括在本发明内。例如,本发明包括与任何人il-37同种型a、b、c、d或e具有同一性的多肽。
使il-37单体二聚化的二聚界面是指参与一个分子与另一个分子结合的两个il-37分子之间的界面。通常,当两个分子相互作用时界面不是溶剂可及的。界面包括进行关键相互作用和/或有助于界面的埋藏表面区域的残基。界面在相互作用中在每个分子上的表面积大小或残基组成方面可以相同或不同。界面包括下表1中列出的任何一个或多个残基,这些残基发生关键相互作用和/或有助于界面的埋藏表面区域。β3-β4环(残基83-91)是控制二聚化的二级结构的关键区域。其他地区也有助于界面。y85压(pack)在由val71、val80和ile78形成的疏水表面上。将y85埋入该表面区域对于二聚体形成来说显得至关重要,因为侧链向丙氨酸的突变消除了二聚体。
表1.列出了有助于il-37二聚体界面的残基。这些中的任何一个或多个是可被遗传/化学修饰以干扰il-37二聚化的残基。数据来源于pdbepisa(embl)。
还考虑降低或抑制其参与特定相互作用类型能力的表1中任何一个或多个残基的突变或修饰。例如,在本发明中包括减少或抑制形成盐桥的能力的asp73处或等价于其的位置处的任何突变。这同样适用于由ser74、asn84、tyr85、ile86和arg87形成的氢键。
β3-β4环(残基83-91)区域的任何修饰都可以将平衡从二聚体转移到单体。这包括但不限于缺失环、缩短环、延长环、突变环的残基和/或化学修饰环残基。在环区域中,lys83、asn84、tyr85、ile86、arg87和pro88看起来是最关键的残基,因此被认为用于突变或修饰。
本发明还提供了抑制il-37多肽二聚化的化合物。优选地,该化合物结合和/或破坏二聚化界面。该化合物可以是与二聚化界面中的至少一个氨基酸残基相互作用的肽、肽模拟物、抗体或小分子。
此外,本发明还提供了抑制il-37多肽二聚化的化合物在制备用于抑制炎症的药物中的用途。而且,本发明提供了抑制有此需要的受试者中的炎症的方法,包括施用抑制il-37多肽二聚化的化合物。优选地,该化合物结合并破坏二聚界面。该化合物可以是与二聚化界面中的至少一个氨基酸残基相互作用的肽、肽模拟物、抗体或小分子。
本发明还提供了肽,其包含、基本上由或由seqidno:1的二聚化界面的氨基酸序列的至少5、6、7、8、9、10、11、12、13、14、15、16、17或18个连续氨基酸组成。优选地,连续氨基酸是seqidno:1的从v71至p88。该肽可用于产生能够抑制il-37多肽二聚化的抗体。
如本文所用,可通过本领域技术人员已知的任何手段来确定与seqidno:1中的位置等价的位置处的氨基酸残基。例如,一个或多个序列与seqidno:1的氨基酸序列的比对将允许本领域技术人员确定在与seqidno:1中的位置等价的位置处的氨基酸。本领域技术人员可以将多肽的三维结构与具有seqidno:1的氨基酸序列的多肽的三维结构进行比较,并确定seqidno:1中的等价位置的氨基酸残基。
如本文所用,“降低的形成二聚体的能力”是指与参考多肽相比,多肽形成同二聚体和/或异二聚体的倾向较小。多肽形成二聚体的能力可以通过各种方法来测量,包括本文所述的那些方法,如尺寸排阻色谱和多角度光散射。优选地,与参考多肽相比,具有降低的形成二聚体的能力的多肽在二聚体形成中具有至少约10%、20%、30%、40%、50%、60%、70%、80%、90%或100%的降低。参考多肽通常会是天然的、野生型的、未修饰的或未突变的多肽。为了评估以给定形式(即单体或二聚体)存在的多肽的比例,合适的方法是例如在非变性条件下的分析凝胶电泳。在这样的方法中,将多肽溶液与一组标准分子量标记一起在聚丙烯酰胺凝胶中跑胶。如果多肽形成二聚体,则在对应于分子量大约是计算的所述多肽氨基酸总量的两倍的物质(species)的凝胶中将观察到蛋白条带。还可以观察到第二条带,其对应于具有计算的所述多肽的氨基酸总和的近似分子量的物质-这代表单体形式的序列。这些条带的相对强度可以用来定量每种形式存在的多肽的比例。类似的方法可以通过替代手段评估分子量,例如分析离心,质谱或尺寸排阻色谱。或者,可使用反相高效液相色谱(rp-hplc)对单体或二聚体进行定量,其中二聚体和更高的低聚物物质基于其疏水性的差异与单体分开。物质的鉴定可以使用质谱检测来实现。同样的方法可进行适应性修改来评估给定的多肽是否表现出异二聚体化或同二聚化的倾向。
本发明多肽的抗炎性质可以通过本文所述的任何方法来确定,特别是在实施例中所述的。
当用于描述本文公开的各种多肽时,“分离的”是指已经从其天然环境的组分中鉴定和分离和/或回收的多肽。其天然环境的污染物成分通常会干扰所述多肽的诊断或治疗用途,并且可包括酶、激素和其他蛋白质的或非蛋白质的溶质。在优选的实施方案中,多肽会被纯化(1)至足以通过使用旋杯式序列分析仪获得至少15个n末端残基或内部氨基酸序列的程度,或(2)至通过sds-page使用考马斯蓝或优选银染的非还原或还原条件下的均匀性。分离的蛋白质包括重组细胞内的原位多肽,因为多肽天然环境的至少一种组分将不存在。然而,通常通过至少一个纯化步骤来制备分离的多肽。
“片段”是本发明多肽的一部分,其保留与多肽基本上相似的功能活性或基本上相同的生物学功能或活性,这可以使用本文所述的测定来确定。
关于多肽序列(即本文定义的本发明的多肽)的“氨基酸序列同一性百分比(%)”或“同一性百分比(%)”被定义为与本发明的特定多肽中的氨基酸残基进行序列比对后候选序列中相同的氨基酸残基的百分比,如果需要引入缺口以实现最大的序列同一性百分比,并且不考虑任何保守置换作为序列同一性的一部分。
本领域技术人员可以确定用于测量比对的合适参数,包括为了在被比较的全长序列上实现最大比对所需的任何算法(以下描述的非限制性实例)。当氨基酸序列比对时,给定氨基酸序列a相对于、与或针对给定氨基酸序列b的氨基酸序列同一性百分比(其可以可替代地表达为给定氨基酸序列a,其具有或包含相对于、与或针对给定氨基酸序列b的某种氨基酸序列同一性百分比)可以如下计算:氨基酸序列同一性百分比=x/y100,其中x是通过a和b的序列比对程序的或算法的比对评分为相同匹配的氨基酸残基的数目,而y是b中氨基酸残基的总数。如果氨基酸序列a的长度不等于氨基酸序列b的长度,则a相对于b的氨基酸序列同一性百分比将不等于b相对于a的氨基酸序列同一性百分比。
在计算同一性百分比时,通常对精确匹配进行计数。两个序列之间的同一性百分比的确定可以使用数学算法来完成。用于比较两个序列的数学算法的非限制性实例是karlin和altschul(1990)proc.natl.acad.sci.usa87:2264的算法,其被修改为如karlin和altschul(1993)proc.natl.acad.sci.usa90:5873-5877中的算法。这样的算法被并入altschul等人(1990)j.mol.biol.215:403的blastn和blastx程序中。为了获得用于比较目的的加空位比对(gappedalignment),可以使用如altschul等人(1997)nucleicacidsres.25:3389中所述的gappedblast(在blast2.0中)。或者,可以使用psi-blast执行迭代搜索,其检测分子之间的远距离关系。参见altschul等人(1997)同上。当使用blast、gappedblast和psi-blast程序时,可以使用各个程序(例如blastx和blastn)的默认参数。也可以通过检查而手动进行比对。用于比较序列的数学算法的另一个非限制性实例是clustalw算法(higgins等人(1994)nucleicacidsres.22:4673-4680)。clustalw比较序列并比对整个氨基酸或dna序列,从而能够提供关于整个氨基酸序列的序列保守的数据。clustalw算法用于几种可商购获得的dna/氨基酸分析软件包,如vectorntiprogramsuite(invitrogencorporation,carlsbad,ca)的alignx模块。用clustalw比对氨基酸序列后,可以评估氨基酸同一性百分比。可用于分析clustalw比对的软件程序的非限制性实例是genedoctm或jalview(http://www.jalview.org/)。genedoctm允许评估多种蛋白质之间的氨基酸(或dna)相似性和同一性。用于比较序列的数学算法的另一非限制性实例是myers和miller(1988)cabios4:11-17的算法。这样的算法被并入到作为gcgwisconsingeneticssoftwarepackage,version10的一部分的align程序(版本2.0)中(可从accelrys,inc.,9685scrantonrd.,sandiego,ca,usa获得)。当使用align程序比较氨基酸序列时,可以使用pam120权重残基表,空位长度罚分为12,空位罚分为4。
多肽有利地包含氨基末端和羧基末端。多肽可以包含d-氨基酸、l-氨基酸或d-和l-氨基酸的混合物。然而,d-氨基酸的形式是特别优选的,因为预期包括d-氨基酸的多肽在体内具有更大的生物活性保留。
多肽可以通过许多常规技术中的任何一种来制备。多肽可以从天然存在的来源或从重组来源分离或纯化。重组生产是优选的。例如,在重组多肽的情况下,可以使用众所周知的分子遗传技术将编码所需肽的dna片段亚克隆到合适的载体中(参见例如maniatis等人,molecularcloning:alaboratorymanual,2nded.(coldspringharborlaboratory,1982);sambrook等人,molecularcloningalaboratorymanual,2nded.(coldspringharborlaboratory,1989))。片段可被转录,随后体外翻译成多肽。可以使用市售试剂盒(例如,由clontech,paloalto,calif.;amershampharmaciabiotechinc.,piscataway,n.j.;invitrogen,carlsbad,calif.等制造的)。聚合酶链式反应可任选地用于操作核酸。
本文使用的术语“保守置换”是指用天然或非天然存在的氨基酸或具有类似空间性质的肽模拟物替换肽中天然序列中存在的氨基酸。当要置换的天然氨基酸的侧链为极性的或疏水性的时候,保守置换应当是用也为极性的或疏水性的(除了具有与被置换的氨基酸的侧链相同的空间性质外)的天然存在的氨基酸、非天然存在的氨基酸或肽模拟物部分。
提供功能上相似的氨基酸的保守氨基酸置换表是本领域普通技术人员熟知的。以下六组是可被认为是彼此的保守置换的氨基酸的实例:
1)丙氨酸(a)、丝氨酸(s)、苏氨酸(t);
2)天冬氨酸(d)、谷氨酸(e);
3)天冬酰胺(n)、谷氨酰胺(q);
4)精氨酸(r)、赖氨酸(k);
5)异亮氨酸(i)、亮氨酸(l)、甲硫氨酸(m)、缬氨酸(v);和
6)苯丙氨酸(f)、酪氨酸(y)、色氨酸(w)。
由于天然存在的氨基酸通常根据其性质分组,所以可以确定天然存在的氨基酸的保守置换,同时考虑到通过空间相似的非带电荷氨基酸替换带电荷的氨基酸被认为是保守置换的事实。为了通过非天然存在的氨基酸产生保守置换,也可以使用本领域众所周知的氨基酸类似物(合成氨基酸)。天然存在的氨基酸的肽模拟物在本领域技术人员已知的文献中有充分记载,非天然或非天然氨基酸在下文中进一步描述。当影响保守置换时,置换氨基酸应在侧链上具有与原始氨基酸相同或相似的官能团。
如本文所用,短语“非保守置换”或“非保守残基”是指通过具有不同电化学和/或空间性质的另一种天然存在或非天然存在的氨基酸置换存在于亲本序列中的氨基酸。因此,置换氨基酸的侧链可以比被置换的天然氨基酸的侧链明显更大(或更小),和/或可以具有与被置换的氨基酸具有显著不同的电子性质的官能团。这种非保守置换的实例包括苯丙氨酸或环己基甲基甘氨酸置换丙氨酸,异亮氨酸置换甘氨酸,或-nh-ch[(-ch2)5-cooh]-co-置换天冬氨酸。非保守置换包括任何不被视为保守的突变。
非保守氨基酸置换可由以下改变引起:(a)置换区域中氨基酸主链的结构;(b)氨基酸的电荷或疏水性;或(c)氨基酸侧链的大小。通常预期产生蛋白质性质的最大改变的置换是下述情形:(a)亲水性残基置换疏水性残基(或被疏水性残基置换);(b)脯氨酸置换任何其他残基(或被任何其他残基置换);(c)具有大体积侧链的残基例如苯丙氨酸置换不具有侧链的残基(或被不具有侧链的残基置换),例如甘氨酸;或(d)具有正电性侧链的残基(例如赖氨酰、精氨酰或组氨酰)置换电负性残基(或被电负性残基置换),例如谷氨酰或天冬氨酰。
可以通过本领域技术人员已知的各种手段来完成例如通过插入、缺失和/或置换来改变天然氨基酸序列以产生突变多肽。例如,可通过将合成的包含修饰位点的寡核苷酸连接到表达载体中来引入位点特异性突变。或者,可以使用寡核苷酸指导的位点特异性诱变方法,如walder等人,gene42:133(1986);bauer等人,gene37:73(1985);craik,biotechniques,12-19(january1995);和美国专利号4,518,584和4,737,462中公开的。用于引入突变的优选方法是quikchangesite-directedmutagenesiskit(stratagene,lajolla,calif.)。
可以使用任何合适的表达载体(例如,如pouwels等人,cloningvectors:alaboratorymanual(elsevier,n.y.:1985)中所述的)和相应的合适的宿主来产生重组多肽。表达宿主包括但不限于埃希氏杆菌属(escherichia)、芽孢杆菌属(bacillus)、假单胞菌属(pseudomonas)、沙门氏菌属(salmonella)内的细菌物种、哺乳动物或昆虫宿主细胞系统(包括杆状病毒系统)(例如,如luckow等人,bio/technology6:47(1988)中所述的),并建立了诸如cos-7、c127、3t3、cho、hela和bhk细胞系等细胞系。本领域技术人员知道表达宿主的选择对所产生的多肽的类型有影响。例如,在酵母或哺乳动物细胞(例如cos-7细胞)中产生的多肽的糖基化将与在细菌细胞如大肠杆菌中产生的多肽的糖基化不同。
或者,本发明的多肽可以使用本领域普通技术人员众所周知的标准肽合成技术来合成(例如,如bodanszky,principlesofpeptidesynthesis(springer-verlag,heidelberg:1984)中概括的)。特别地,多肽可以使用固相合成法来合成(参见,例如,merrifield,j.am.chem.soc.85:2149-54(1963);barany等人,int.j.peptideproteinres.30:705-739(1987);和美国专利号5,424,398)。如果需要,可以使用自动肽合成仪完成。去除叔丁氧基羰基(t-boc)或9-芴基甲氧基羰基(fmoc)氨基酸封闭基团并从树脂中分离多肽可以通过例如在降低的温度下进行酸处理来完成。然后可以将含有多肽的混合物用例如二甲醚萃取以去除非肽类有机化合物,并且可以从树脂粉末中(例如用约25%w/v的乙酸)提取合成的多肽。在多肽合成之后,可以任选进行进一步的纯化(例如使用高效液相色谱(hplc))以消除任何不完全的多肽或游离氨基酸。可以对合成的多肽进行氨基酸和/或hplc分析以验证其身份。对于根据本发明的其它应用,产生作为较大融合蛋白的一部分的多肽可能是优选的,例如通过本文所述的方法或其他遗传手段,或者作为更大的缀合物的一部分,如通过物理或化学方法缀合,如本领域普通技术人员已知并在此描述的。
本发明的多肽还可以通过与另一部分缀合或融合来修饰,以促进纯化,或增加多肽的体内半衰期,或用于使用本领域已知的方法的免疫测定。例如,本发明的多肽可以通过糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过已知的保护/封闭基团衍生化、蛋白酶解切割、与细胞配体或其他蛋白质的连接等进行修饰。
“肽模拟物”是具有与本发明的多肽基本相同的结构和/或功能特征的合成化学化合物,前者在本文中进一步描述。典型地,肽模拟物具有与本发明的多肽相同或相似的结构,例如具有降低的形成二聚体能力的seqidno:1的相同或相似序列或其片段。肽模拟物通常含有至少一个非天然合成的残基。肽模拟物化合物的非天然组分可以是根据以下中的一种或多种:a)天然酰胺键(“肽键”)键以外的残基连接基团;b)代替天然存在的氨基酸残基的非天然残基;或c)诱导二级结构模拟的残基,即诱导或稳定二级结构,例如β转角、γ转角、β片层、α螺旋构象等。
肽模拟物可以使用科学和专利文献中描述的各种程序和方法合成,例如organicsynthesescollectivevolumes,gilman等人(eds)johnwiley&sons,inc.,ny,al-obeidi(1998)mol.biotechnol.9:205-223;hruby(1997)curr.opin.chem.biol.1:114-119;ostergaard(1997)mol.divers.3:17-27;ostresh(1996)methodsenzymot.267:220-234。
本文考虑的修饰包括但不限于修饰侧链,在多肽合成过程中掺入非天然氨基酸和/或其衍生物,以及使用交联剂和对本发明的多肽施加构象约束的其它方法。本文考虑了降低分子形成二聚体的能力的任何修饰,包括翻译后修饰。实例包括本领域中已知的通过点击化学掺入的修饰。示例性修饰包括聚乙二醇化和糖基化。
本发明所考虑的侧链修饰的实例包括氨基的修饰,例如通过与醛反应进行还原性烷基化,接着用nabh4还原;用甲基乙酰亚胺酯进行脒化;用乙酸酐酰化;用氰酸酯氨基甲酰化氨基;用2,4,6-三硝基苯磺酸(tnbs)三硝基苄基化氨基;用琥珀酸酐和四氢邻苯二甲酸酐酰化氨基;和用吡哆醛-5-磷酸盐对赖氨酸进行吡哆醛化,接着用nabh4还原。
精氨酸残基的胍基可以通过与试剂如2,3-丁二酮、苯基乙二醛和乙二醛形成杂环缩合产物而被修饰。
羧基可以通过经由o-酰基异脲形成的碳化二亚胺活化,接着随后衍生为例如相应的酰胺而被修饰。
巯基可以通过如用碘乙酸或碘乙酰胺羧甲基化的方法进行修饰;过甲酸氧化成磺基丙氨酸;与其他硫醇化合物形成混合二硫化物;与马来酰亚胺、马来酸酐或其它置换的马来酰亚胺反应;使用4-氯汞苯甲酸、4-氯汞苯磺酸、苯基氯化汞、2-氯汞-4-硝基苯酚和其他汞制剂形成汞衍生物;在碱性ph下用氰酸酯进行氨基甲酰化。
色氨酸残基可通过例如用n-溴代琥珀酰亚胺氧化或用2-羟基-5-硝基苄基溴或磺苯基卤化物使吲哚环烷基化来修饰。另一方面,可以通过用四硝基甲烷硝化改变酪氨酸残基,以形成3-硝基酪氨酸衍生物。
组氨酸残基的咪唑环的修饰可以通过用碘乙酸衍生物进行烷基化或用焦碳酸二乙酯进行n-乙氧羰基化来完成。
在蛋白质合成期间掺入非天然氨基酸和衍生物的实例包括但不限于使用正亮氨酸、4-氨基丁酸、4-氨基-3-羟基-5-苯基戊酸、6-氨基己酸、叔丁基甘氨酸、正缬氨酸、苯基甘氨酸、鸟氨酸、肌氨酸、4-氨基-3-羟基-6-甲基庚酸、2-噻吩基丙氨酸和/或氨基酸的d-异构体。表2中显示了本文考虑的非天然氨基酸列表。
表2
通过使用同双功能交联剂,例如具有n=1至n=6的(ch2)n间隔基团的双功能酰亚胺酯、戊二醛、n-羟基琥珀酰亚胺酯和通常含有氨基反应性部分例如n-羟基琥珀酰亚胺和另一个基团特异性反应性部分的异双功能试剂,交联剂可用于例如稳定3d构象。
编码本发明的任何多肽的核酸分子也在本发明的范围内。核酸可用于例如制备本发明的多肽和作为治疗剂。它们可以施用于在培养物中的或体内的细胞,并且可以包括指导或促进从细胞分泌本发明多肽的分泌信号。同样在本发明的范围内的是包含或包括本发明的核酸(下面进一步描述)的表达载体和宿主细胞。尽管根据定义,本发明的核酸可以被称为“分离的”,但是本发明的多肽不是野生型多肽,并且因此不会被天然存在的核酸编码。因此,虽然本发明的多肽和核酸可以是“纯化的”、“基本上纯化的”、“分离的”、“重组的”或“合成的”,但是它们不一定要这样以与天然存在的材料区分。
“分离的”核酸分子是从至少一种污染性核酸分子中鉴定和分离的核酸分子,所述污染性核酸分子在编码多肽(通常为il-37)的核酸的天然来源中通常相关。分离的核酸分子不是处于在自然界中发现的形式或环境。因此,分离的核酸分子与存在于天然细胞中的核酸分子不同。然而,分离的核酸分子包括通常表达il-37的细胞中含有的核酸分子,例如,其中该核酸分子位于与天然细胞不同的染色体位置。
术语“核酸分子”和“多核苷酸”在本文中可互换使用,并且是指任何长度的核苷酸(脱氧核糖核苷酸或核糖核苷酸或其类似物)的聚合形式。多核苷酸的非限制性实例包括基因、基因片段、信使rna(mrna)、cdna、重组多核苷酸、质粒、载体、任何序列的分离的dna、任何序列的分离的rna、核酸探针和引物。本发明的多核苷酸可以分离或纯化形式提供。“编码”选定多肽的核酸序列是当置于合适的调节序列控制下时体内转录(在dna的情况下)并翻译(在mrna的情况下)转化成多肽的核酸分子。编码序列的边界由5'(氨基)末端的起始密码子和3'(羧基)末端的翻译终止密码子决定。为了本发明的目的,这样的核酸序列可以包括但不限于来自病毒、原核或真核mrna的cdna,来自病毒或原核dna或rna的基因组序列,甚至合成的dna序列。转录终止序列可以位于编码序列的3'。
本发明的多核苷酸可根据本领域众所周知的方法合成,例如sambrook等人(1989,molecularcloning—alaboratorymanual;coldspringharborpress)中所述的。
本发明的多核苷酸分子可以表达盒的形式提供,所述表达盒包括与插入的序列有效连接的控制序列,从而允许本发明的多肽在靶向受试者体内表达。这些表达盒进而通常在适合用作核酸免疫的试剂的载体(例如,质粒或重组病毒载体)内提供。这样的表达盒可以直接施用于宿主受试者。或者,可以将包含本发明的多核苷酸的载体施用于宿主受试者。优选地,使用遗传载体来制备和/或施用多核苷酸。合适的载体可以是能够携带足够量的遗传信息并允许表达本发明的多肽的任何载体。
因此,本发明包括包含这种多核苷酸序列的表达载体。因此,本发明提供了用于预防或治疗炎性疾病或病症的载体,所述载体包含编码本发明多肽的多核苷酸序列和任选地编码如本文所定义的不同多肽的一个或多个另外的多核苷酸序列。
此外,应该理解,本发明的组合物和产品可以包含多肽和多核苷酸的混合物。因此,本发明提供了如本文所定义的组合物或产品,其中能够表达所述多肽的多核苷酸代替任何一种多肽。
表达载体在分子生物学领域中常规构建,并且可以例如涉及使用质粒dna和合适的起始子,启动子,增强子和其他元件,例如可能需要的聚腺苷酸化信号,并且其位于正确的取向,以允许表达本发明的肽。其他合适的载体对于本领域技术人员来说是显而易见的。就此而言,我们例如参考sambrook等人。
因此,本发明的多肽可以通过将这种载体递送至细胞并允许从载体转录而提供。优选地,本发明的多核苷酸或用在本发明的载体中的多核苷酸有效地连接至能够使宿主细胞表达编码序列的控制序列,即载体是表达载体。
“有效地连接”指的是其中如此描述的组分被配置以执行其通常功能的元件的布置。因此,当存在适当的酶时,与核酸序列有效连接的给定调控序列(例如启动子)能够影响该序列的表达。启动子不需要与序列连续,只要它起着指导其表达的作用即可。因此,例如,在启动子序列和核酸序列之间可以存在插入的非翻译但转录的序列,并且启动子序列仍然可以被认为与编码序列“有效地连接”。
本领域已经描述了许多表达系统,其中的每一个均通常由含有有效地连接至表达控制序列的目的基因或核苷酸序列的载体组成。这些控制序列包括转录启动子序列和转录起始和终止序列。本发明的载体可以是例如提供有复制起点,任选的用于表达所述多核苷酸的启动子和任选的启动子的调节子,的质粒,病毒或噬菌体载体。“质粒”是染色体外遗传元件形式的载体。载体可以含有一个或多个选择标记基因,例如在细菌质粒或真菌载体的抗性基因的情况下为氨苄青霉素抗性基因。载体可以在体外使用,例如用于生产dna或rna或用于转染或转化宿主细胞,例如哺乳动物宿主细胞。载体也可以适合在体内使用,例如允许体内表达多肽。
“启动子”是起始和调控编码多肽的多核苷酸的转录的核苷酸序列。启动子可以包括诱导型启动子(其中有效地连接到启动子的多核苷酸序列的表达由被分析物、辅因子、调控蛋白等诱导),阻遏型启动子(其中有效地连接到启动子的多核苷酸序列的表达由被分析物、辅因子、调节蛋白等阻遏)和组成型启动子。术语“启动子”或“控制元件”意图包括这些区域的全长启动子区域和功能性(例如,控制转录或翻译)区段。
根据本发明的多核苷酸、表达盒或载体可以另外包含信号肽序列。通常将信号肽序列插入到与启动子有效连接的位置,使得信号肽被表达并促进也由与启动子有效地连接的编码序列编码的多肽的分泌。
通常,信号肽序列编码10至30个氨基酸,例如15至20个氨基酸的肽。通常这些氨基酸主要是疏水的。在典型情况下,信号肽将携带信号肽的生长多肽链靶向表达细胞的内质网。信号肽在内质网中被切除,允许通过高尔基体分泌多肽。因此,本发明的肽可以通过从个体内的细胞表达和从那些细胞分泌而提供给个体。
短语“治疗有效量”通常指本发明的一种或多种多肽或多核苷酸的量,其(i)治疗特定的疾病,病症或障碍,(ii)减弱,改善或消除特定的疾病,病症或障碍的一种或多种症状,或(iii)延迟本文所述的特定疾病,病症或障碍的一种或多种症状的发作。
本发明可用于治疗各种炎性疾病或病症。如本文所用,“炎性疾病或病症”包括急性或慢性炎症和炎性障碍,例如与自身免疫病相关的炎症、心血管炎症(例如动脉粥样硬化、中风)、胃肠炎症、肝炎症障碍、肺部炎症(例如哮喘、呼吸机诱导的肺损伤)、肾脏炎症、眼部炎症(例如,葡萄膜炎)、胰腺炎症、泌尿生殖器炎症、神经炎性障碍(例如多发性硬化、阿尔茨海默氏病)、变态反应(例如变应性鼻炎/鼻窦炎、皮肤变态反应和障碍(例如荨麻疹(urticaria/hives)、血管性水肿、特应性皮炎、接触性皮炎、牛皮癣)、食物过敏、药物过敏、昆虫过敏、肥大细胞增多症)、骨骼炎症(例如关节炎、骨关节炎、类风湿性关节炎、脊柱关节病)、感染(例如细菌或病毒感染;口腔炎症障碍(即牙周炎(perodontis)、牙龈炎或口炎(somatitis));和移植(例如,同种异体移植或异种移植排斥、母体-胎儿耐受、移植物抗宿主疾病)。
本发明可用于治疗建议用重组野生型il-37治疗的各种炎性疾病或病症,例如dinarello等人eur.j.immunol.(2016)46:1067-1081,特别是表3提及的疾病或病症。
考虑在本发明的方法或应用中,或通过本发明的多肽或药物组合物治疗的炎性疾病或病症包括主要由toll样受体(tlr)2、4、7、8和/或9活化介导的那些。与tlr活化有关的那些疾病和病症的概述可见于connolly等人currentopinioninpharmacology2012,12:510-518中的图1和表1中。预期通过本发明的方法或用途或通过本发明的多肽或药物组合物以及主要介导该适应症的炎症的tlr治疗的示例性适应症为:
·缺血/再灌注损伤、心脏缺血、移植物功能延迟(tlr2);
·类风湿性关节炎(tlr2);
·系统性红斑狼疮(tlr7、8和/或9);
·脓毒症(tlr4和其他tlr);和
·急性和慢性炎症(tlr4)。
本发明的方法或用途或者本发明的多肽或药物组合物可用于减少、抑制或预防由其他介质例如其他细胞因子(il-1、ifnγ等)、其它介质(例如补体、白三烯等)以及化学和物理损伤诱导的炎症。
自身免疫病包括例如获得性免疫缺陷综合征(aids,其为具有自身免疫性组分的病毒性疾病)、斑秃、强直性脊柱炎、抗磷脂综合征、自身免疫性艾迪生病、自身免疫性溶血性贫血、自身免疫性肝炎、自身免疫性内耳疾病(aied)、自身免疫性淋巴组织增生综合征(alps)、自身免疫性血小板减少性紫癜(atp)、白塞氏病、心肌病、口炎性腹泻(celiacsprue)-疱疹样皮炎;慢性疲劳免疫功能紊乱综合征(cfids)、慢性炎性脱髓鞘性多发性神经病(cipd)、瘢痕性类天疱疮、冷凝集素病、crest综合征、克罗恩病、恶性萎缩性丘疹病、青少年皮肌炎、盘状狼疮、原发性冷球蛋白血症、纤维肌痛、格雷夫斯病、吉兰-巴雷综合征、桥本甲状腺炎、特发性肺纤维化、特发性血小板减少性紫癜(itp)、iga肾病、胰岛素依赖性糖尿病、青少年慢性关节炎(斯提耳氏病)、青少年类风湿性关节炎、美尼尔氏病、混合性结缔组织病、多发性骨髓瘤、重症肌无力、恶性贫血(pemaciousanemia)、结节性多动脉炎、多软骨炎、多腺性综合征、风湿性多肌痛、多发性肌炎和皮肌炎、原发性无丙种球蛋白血症、原发性胆汁性肝硬化、牛皮癣、牛皮癣性关节炎、雷诺氏现象、莱特尔氏综合征、风湿热、类风湿性关节炎、结节病、硬皮病(进行性系统性硬化病(pss),也称为系统性硬化病(ss))、干燥综合征、僵人综合征、系统性红斑狼疮、大动脉炎、颞动脉炎/巨细胞动脉炎、溃疡性结肠炎、葡萄膜炎、白癜风和韦格纳氏肉芽肿病。
优选地,炎性疾病或病症是多发性硬化、类风湿性关节炎、皮肤过敏如特应性皮炎、接触性皮炎、牛皮癣、炎症性肠病、葡萄膜炎、干眼病、系统性硬化症(硬皮病)、牙周病、白癜风、sle/盘状狼疮/格雷夫病、动脉粥样硬化、哮喘或迟发型超敏反应。
多发性硬化(ms)是一种涉及大脑和脊髓轴突周围的髓鞘脱髓鞘的炎性疾病。ms症状包括但不限于脑和/或脊髓中的白质疤痕和各种各样的神经症状,包括但不限于感觉的改变,例如丧失敏感性或麻刺感、刺痛感或麻木(知觉下降和麻痹)、肌肉无力、阵挛、肌肉痉挛或移动困难;协调和平衡方面的困难(共济失调);发音(构音障碍)或吞咽(吞咽困难)问题,视觉问题(眼球震颤、视神经炎等),疲劳,急性/慢性疼痛以及膀胱和肠道困难。不同程度的认知障碍和抑郁症也很常见。ms的症状通常出现在神经功能逐渐进展性恶化的偶发性急性恶化期,或两者兼而有之。
类风湿性关节炎是一种慢性全身性炎性疾病,其可能影响许多组织和器官,但主要是攻击滑膜关节。该过程涉及滑膜细胞增生,滑液过多和滑膜纤维组织发展继发的关节周围的滑膜囊的炎症反应。疾病过程的病理学常常导致关节软骨的破坏和关节强直。类风湿性关节炎也可以在肺、心包膜、肺胸膜、巩膜和结节性病变中产生弥漫性炎症,最常见于皮下组织。
其他炎性疾病或病症包括与中风、心肌梗塞、缺血再灌注损伤和移植相关的急性炎症。此外,慢性炎症与自身炎性疾病或自身免疫病如上述多发性硬化和糖尿病(1型或2型糖尿病)相关。
进一步的炎性疾病或病症是脓毒症、脓毒性休克或内毒素休克。脓毒症或内毒素休克可能由细菌、真菌、病毒或寄生虫引起。来自细菌的致病因素可以是脂多糖(lps),也称为脂聚糖和内毒素,其是由脂质和由o-抗原组成的多糖组成的大分子,外核和内核通过共价键连接;它们存在于革兰氏阴性细菌的外膜中,并在动物体内引起强烈的免疫反应。
对于本文所述的任何炎性疾病或病症,当将本发明的多肽局部施用于人时,化合物的治疗有效量优选对应于约0.01至约10%(w/w),或者约0.1至10%(w/w),或约1.0至约10%(w/w),约0.1至约5%(w/w),或约1.0至约5%(w/w)。在本文所述的任何炎性疾病或病症中,当将本发明的多肽口服施用于受试者时,化合物的治疗有效量优选对应于约1至约50mg/kg,或约1至约25mg/kg,或约1至约10mg/kg,约5至约25mg/kg,或约10至约20mg/kg之间。
在本发明的组合物中,作为单体存在的本发明多肽的比例可以为存在于组合物中的总多肽的至少约0.5%、1%、2%、3%、4%、5%、6%、7%、8%约9%、10%、20%、30%、40%、50%、60%、70%、80%或90%,通常在合适条件下在溶液中存储合适的时间段。合适的时间段和条件包括本领域技术人员在使用之前合理地期望使多肽保持在溶液中的时间范围和条件。例如,约24小时、约48小时或约72小时的时间段是典型的,尽管一些溶液可以保持较长时间,例如至少一周、一个月、6个月、1年、2年、3年或更长时间。存储条件通常可以是室温和相对潮湿,或者通常为25℃和60%的相对湿度,但是可以包括本领域技术人员遇到的任何标准储存条件,例如约4℃、-20℃、或-80℃。
施用的频率可以是每天一次,或者每天2次或3次。治疗期可以是可检测疾病的持续时间。
典型地,配制治疗有效剂量以包含至少约0.1%直至约50%或更高的浓度(以重量计),以及其中范围的所有组合和子组合。该组合物可以配制成含有本发明的一种或多种多肽,其浓度为从约0.1至小于约50%,例如约49、48、47、46、45、44、43、42、41或40%,其浓度从大于约0.1%,例如约0.2、0.3、0.4或0.5%,至小于约40%,例如约39、38、37、36、35、34、33、32、31或30%。示例性组合物可含有从约0.5%至小于约30%,例如约29、28、27、26、25、25、24、23、22、21或20%,其中浓度为从大于约0.5%,例如约0.6、0.7、0.8、0.9或1%,至小于约20%,例如约19、18、17、16、15、14、13、12、11或10%。组合物可以含有从大于约1%,例如约2%,至小于约10%,例如约9或8%,包括大于约2%,例如约3或4%,到小于约8%,例如约7或6%的浓度。活性剂可以例如以约5%的浓度存在。在所有情况下,可以调整这些量以补偿实际递送至处理的细胞或组织的活性成分的量的差异。
尽管本发明用于人类,但是本发明也可用于治疗兽医目的。本发明对于家畜动物或农场动物如牛、羊、马和家禽;对于伴侣动物如猫和狗;和对于动物园的动物是有用的。
药物组合物可以配制用于任何适当的施用途径,包括例如外用(例如透皮或眼睛)、经口、经颊、经鼻、经阴道、经直肠或肠胃外施用。本文使用的术语“肠胃外”包括皮下、皮内、血管内(例如静脉内)、肌内、脊髓、颅内、鞘内、眼内、眼周、眼眶内、滑膜内和腹膜内注射以及任何类似的注射或输注技术。在某些实施方案中、适于口服或肠胃外使用的形式的组合物是优选的。合适的口服形式包括例如片剂、糖锭剂(troche)、锭剂(lozenge)、水性或油性混悬剂、可分散的粉剂或颗粒剂、乳液、硬或软胶囊剂或糖浆剂或酏剂。在其他实施方案中,本文提供的组合物可以配制成冻干物。
各种剂量单位各自优选作为分立剂量片剂、胶囊、锭剂、糖衣丸、胶剂或其他类型的固体制剂提供。胶囊可以封装粉末、液体或凝胶。该固体制剂可以被吞咽,或者可以是可吮吸型或可咀嚼型(易碎的或胶状的)。本发明考虑了不同于泡罩包装的剂量单位保持装置;例如包装,例如瓶、管、罐、小包。剂量单位还可以包括药物制剂实践中众所周知的常规赋形剂,如粘合剂、胶凝剂、填充剂、压片润滑剂、崩解剂、表面活性剂和着色剂;和可吮吸或可咀嚼的配方。
意图用于口服使用的组合物可进一步包含一种或多种组分,例如甜味剂、调味剂、着色剂和/或防腐剂,以提供吸引人的和可口的制剂。片剂含有与适用于制造片剂的生理上可接受的赋形剂混合的活性成分。这样的赋形剂包括例如惰性稀释剂如碳酸钙、碳酸钠、乳糖、磷酸钙或磷酸钠,制粒剂和崩解剂如玉米淀粉或海藻酸,粘合剂如淀粉、明胶或阿拉伯胶和润滑剂如硬脂酸镁、硬脂酸或滑石粉。片剂可以是未包衣的,或者它们可以用已知技术包衣以延迟在胃肠道中的崩解和吸收,从而提供长时间的持续作用。例如,可以使用延时材料如甘油单硬脂酸酯或甘油二硬脂酸酯。
用于口服使用的制剂也可以呈现为硬明胶胶囊,其中活性成分与惰性固体稀释剂例如碳酸钙、磷酸钙或高岭土混合,或者作为软明胶胶囊,其中活性成分与水或油介质如花生油、液体石蜡或橄榄油混合。
含水悬浮液含有活性成分与适合制造水悬浮液的赋形剂的混合物。这类赋形剂包括悬浮剂如羧甲基纤维素钠、甲基纤维素、羟丙基甲基纤维素、海藻酸钠、聚乙烯吡咯烷酮、黄蓍胶和阿拉伯胶,以及分散剂或润湿剂如天然存在的磷脂(例如卵磷脂)、亚烷基氧化物与脂肪酸的缩合产物如聚氧乙烯硬脂酸酯、环氧乙烷与长链脂肪醇的缩合产物如十七碳乙烯氧基鲸蜡醇、环氧乙烷与衍生自脂肪酸和己糖醇的偏酯的缩合产物如聚氧乙烯山梨醇单油酸酯或环氧乙烷与衍生自脂肪酸和己糖醇酐的偏酯的缩合产物如聚乙烯脱水山梨糖醇单油酸酯。水悬浮液还可以包含一种或多种防腐剂,例如对羟基苯甲酸乙酯或正丙酯,一种或多种着色剂,一种或多种调味剂和一种或多种甜味剂,如蔗糖或糖精。
油性悬浮液可以通过将活性成分悬浮在植物油如花生油、橄榄油、芝麻油或椰子油中,或悬浮于矿物油如液体石蜡中来配制。油性悬浮液可以含有增稠剂如蜂蜡、硬石蜡或鲸蜡醇。可加入如上所述的甜味剂和/或调味剂以提供可口的口服制剂。这种悬浮液可以通过添加抗氧化剂如抗坏血酸来保存。
适用于通过加水制备含水悬浮液的可分散粉剂和颗粒剂提供了与分散剂或润湿剂,悬浮剂和一种或多种防腐剂混合的活性成分。合适的分散剂或润湿剂和悬浮剂以上面已经提到的那些为例。另外的赋形剂,例如甜味剂,调味剂和着色剂也可以存在。
药物组合物也可以是水包油乳液的形式。油相可以是植物油如橄榄油或花生油、矿物油如液体石蜡或其混合物。合适的乳化剂包括天然存在的胶例如阿拉伯胶或黄蓍胶,天然存在的磷脂例如大豆卵磷脂,以及衍生自脂肪酸和己糖醇的酯或偏酯,酸酐例如脱水山梨糖醇单油酸酯,和衍生自脂肪酸的偏酯和己糖醇与环氧乙烷的缩合产物,如聚氧乙烯脱水山梨糖醇单油酸酯。乳液还可以包含一种或多种甜味剂和/或调味剂。
糖浆和酏剂可以用甜味剂如甘油、丙二醇、山梨糖醇或蔗糖配制。这样的制剂还可以包含一种或多种缓和剂,防腐剂,调味剂和/或着色剂。
本发明的多肽可以配制用于局部或外用(localortopical)给药,例如外部应用于皮肤。用于外部施用的制剂典型地包含与(一种或多种)活性剂组合的外用媒介物,有或没有额外的任选组分。
合适的外用媒介物和附加组分在本领域中是众所周知的,并且显而易见的是,媒介物的选择将取决于具体的物理形式和递送方式。外用媒介物包括有机溶剂如醇(例如乙醇、异丙醇或甘油),二醇如丁二醇、异戊二醇或丙二醇,脂族醇如羊毛脂,水和有机溶剂的混合物以及有机溶剂如醇和甘油的混合物,基于脂质的物质如脂肪酸,酰基甘油包括如矿物油等油类,天然或合成来源的脂肪,磷酸甘油酯、鞘脂类和蜡类,基于蛋白质的物质如胶原、明胶,基于硅酮的物质(非挥发性和挥发性)以及基于烃的材料如微海绵和聚合物基质。
组合物可以进一步包含一种或多种适于改善所施用制剂的稳定性或有效性的组分,例如稳定剂、悬浮剂、乳化剂、粘度调节剂、胶凝剂、防腐剂、抗氧化剂、皮肤渗透促进剂、保湿剂和缓释材料。martindale–theextrapharmacopoeia(pharmaceuticalpress,london1993)和martin(ed.),remington'spharmaceuticalsciences中描述了这些组分的例子。制剂可以包含微胶囊,例如羟甲基纤维素或明胶微胶囊、脂质体、白蛋白微球体、微乳液、纳米颗粒或纳米胶囊。
可以各种物理形式制备外用制剂,包括例如固体剂、糊剂、乳膏剂、泡沫剂、洗剂、凝胶剂、粉剂、含水液体剂、乳液剂、喷雾剂和皮肤贴剂。这样的形式的物理外观和粘度可以由制剂中存在的一种或多种乳化剂和一种或多种粘度调节剂的存在和量来控制。固体剂通常是牢固的且不可倾倒的,并且通常被配制成条或棒,或者以颗粒形式。固体剂可以是不透明或透明的,并且任选地可以包含溶剂、乳化剂、保湿剂、润滑剂、香料、染料/着色剂、防腐剂和增加或增强最终产品功效的其它活性成分。乳膏剂和洗剂通常彼此相似,主要是粘度不同。洗剂和乳膏剂二者可以是不透明的、半透明的或透明的,并且通常含有乳化剂、溶剂和粘度调节剂,以及保湿剂,润滑剂、香料、染料/着色剂、防腐剂和增加或增强最终效果的其它活性成分产品。凝胶剂可以制备一系列的粘度,从稠或高粘度到稀或低粘度。这些制剂,如洗剂和乳膏剂,也可以含有溶剂、乳化剂、保湿剂、润滑剂、香料、染料/着色剂、防腐剂和增加或增强最终产品功效的其它活性成分。液体比乳膏剂、洗剂或凝胶剂更稀,通常不含乳化剂。液体外用产品通常含有溶剂、乳化剂、保湿剂、润滑剂、香料、染料/着色剂、防腐剂和增加或增强最终产品功效的其它活性成分。
用于外用制剂的乳化剂包括但不限于离子乳化剂,鲸蜡硬脂醇,非离子乳化剂如聚氧乙烯油基醚、peg-40硬脂酸、ceteareth-12、ceteareth-20、ceteareth-30、ceteareth醇、peg-100硬脂酸和硬脂酸甘油酯。合适的粘度调节剂包括但不限于保护性胶体或非离子胶如羟乙基纤维素,黄原胶,硅酸镁铝,二氧化硅,微晶蜡,蜂蜡,石蜡和十六烷基棕榈酸酯。可通过加入胶凝剂如壳聚糖、甲基纤维素、乙基纤维素、聚乙烯醇、聚季铵盐、羟乙基纤维素、羟丙基纤维素、羟丙基甲基纤维素、卡波姆或氨化甘草酸盐形成凝胶剂组合物。合适的表面活性剂包括但不限于非离子、两性、离子和阴离子表面活性剂。例如,聚二甲基硅氧烷共聚醇、聚山梨酯20、聚山梨酯40、聚山梨酯60、聚山梨酯80、月桂酰胺dea、椰油酰胺dea和椰油酰胺mea、油基甜菜碱、椰油酰胺丙基磷脂酰pg-二甲基氯化铵和月桂醇聚醚硫酸铵中的一种或多种可用于外用制剂。
防腐剂包括但不限于抗微生物剂如对羟基苯甲酸甲酯、对羟基苯甲酸丙酯、山梨酸、苯甲酸和甲醛,以及物理稳定剂和抗氧化剂如维生素e、抗坏血酸钠/抗坏血酸和没食子酸丙酯。合适的保湿剂包括但不限于乳酸和其它羟基酸及其盐、甘油、丙二醇和丁二醇。合适的润滑剂包括羊毛脂醇、羊毛脂、羊毛脂衍生物、胆固醇、凡士林、新戊酸异硬脂酸酯和矿物油。合适的香料和颜色包括但不限于fd&credno.40和fd&cyellowno.5。可以包含在外用制剂中的其它合适的附加成分包括但不限于研磨剂、吸收剂、抗结块剂、消泡剂、抗静电剂、收敛剂(如金缕梅)、醇和草药提取物如甘菊提取物、粘合剂/赋形剂、缓冲剂、螯合剂、成膜剂、增稠剂、喷射剂、遮光剂、ph调节剂和保护剂。
用于外用组合物的典型递送方式包括使用手指的敷用,使用物理涂药器例如布、纸巾、拭子、棒或刷的敷用,包括雾、气雾剂或泡沫喷雾的喷雾,滴管敷用,喷洒,浸泡和漂洗。还可以使用控释媒介物,并且可以配制用于经皮施用的组合物(例如,作为透皮贴剂)。
药物组合物可以配制成吸入制剂,包括喷雾剂、雾剂或气雾剂。这对于治疗某些炎性疾病或病症可能是特别优选的。对于吸入制剂,本文提供的组合物或组合可以经由本领域技术人员已知的任何吸入方法递送。这样的吸入方法和装置包括但不限于具有喷射剂如cfc或hfa的定量吸入器或生理上和环境上可接受的喷射剂。其他合适的装置是呼吸操作吸入器、多剂量干粉吸入器和气雾剂喷雾器。用于本方法的气雾剂制剂通常包括喷射剂、表面活性剂和共溶剂,并且可以填充到通过合适的计量阀关闭的常规气雾剂容器中。
吸入组合物可以包含含有适于雾化和支气管内使用的活性成分的液体或粉末组合物,或通过分配计量剂量的气雾剂单位给药的气雾剂组合物。合适的液体组合物包含在药学上可接受的水性吸入溶剂如等渗盐水或抑菌水中的活性成分。溶液通过泵或挤压驱动的雾化喷雾分配器或通过任何其他常规手段施用,以导致或使所需剂量的液体组合物吸入患者的肺中。其中载体为液体的用于施用的合适制剂,作为例如鼻喷雾剂或作为滴鼻剂,包括活性成分的水性或油性溶液。
药物组合物也可以栓剂的形式制备,例如用于直肠施用。这样的组合物可以通过将药物与合适的非刺激性赋形剂混合来制备,所述赋形剂在常温下为固体,但在直肠温度下为液体,并因此在直肠中融化以释放药物。合适的赋形剂包括例如可可脂和聚乙二醇。
药物组合物可以配制成缓释制剂,例如在施用后产生调节剂缓慢释放的胶囊。通常可以使用众所周知的技术制备这样的制剂,并通过例如经口,经直肠或经皮下植入,或通过植入期望的靶部位来施用。在这种制剂中使用的载体是生物相容的,并且也可以是生物可降解的。优选地,制剂提供相对恒定水平的调节剂释放。包含在缓释制剂中的调节剂的量取决于例如植入部位、释放的速率和预期的持续时间以及待治疗或预防的病症的性质。
在另一个实施方案中,提供了包括如上所述的本发明的一种或多种多肽或多核苷酸和/或药物组合物的试剂盒或制品。
在其他实施方案中,提供了用于上述治疗或预防应用的试剂盒,所述试剂盒包括:
-容纳本发明的多肽、多核苷酸或药物组合物的容器;
-带有使用说明的标签或包装插页。
在某些实施方案中,试剂盒可以含有一种或多种用于治疗炎性疾病或病症的其它有效物质或成分。
试剂盒或“制品”可以包括容器以及在容器上或与容器相关联的标签或包装插页。合适的容器包括例如瓶、小瓶、注射器、泡罩包装等。容器可以由各种材料形成,例如玻璃或塑料。容器容纳有效治疗病症的治疗组合物,并且可具有无菌进入端口(例如容器可以是静脉内溶液袋或具有能够被皮下注射针头刺穿的塞子的小瓶)。标签或包装插页指出治疗组合物用于治疗选择的病症。在一个实施方案中,标签或包装插页包括使用说明书,并指出治疗或预防组合物可用于治疗本文所述的炎性疾病或病症。
试剂盒可以包含(a)治疗或预防组合物;和(b)其中含有第二有效物质或成分的第二容器。本发明该实施方案中的试剂盒可以进一步包含指示可以使用组合物和其它活性成分来治疗病症或预防源自本文所述的炎性疾病或病症的并发症的包装插页。或者或另外,试剂盒可以进一步包含第二(或第三)容器,所述容器包含药学上可接受的缓冲液,如注射用抑菌水(bwfi)、磷酸盐缓冲盐水、林格氏溶液和葡萄糖溶液。它可以进一步包括从商业和用户观点来看所需的其他材料,包括其他缓冲液、稀释剂、过滤器、针头和注射器。
在某些实施方案中,治疗组合物可以一次性或可重复使用的装置的形式提供,包括用于容纳治疗剂、预防剂或药物组合物的容器。在一个实施方案中,该装置是注射器。该装置可容纳1-2ml的治疗组合物。治疗性或预防性组合物可以即用的状态或以需要混合或添加其他组分的状态提供在装置中。
本发明的多肽或组合物可以用作用于可植入材料和装置例如支架的抗炎涂层。本发明的多肽或组合物可以涂布在可植入材料或装置上或与其一体化。本发明的多肽或组合物可以是聚合物涂层的一部分。
应该理解的是,在本说明书中公开和限定的本发明延伸到从文本或附图中提到或明显可见的两个或更多个单独特征的所有替代组合。所有这些不同的组合构成本发明的各种替代方面。
实施例
实施例1
克隆和蛋白质纯化
将密码子优化的il-37(46-218)克隆到改造的烟草蚀刻病毒蛋白酶可切割形式的pgex-4t-1(gehealthcare)中(7)。在18℃下通过iptg诱导在bl21-codonplus(de3)-ril细胞(stratagene)中表达重组蛋白质。通过在具有两种完全无edta的蛋白酶抑制剂片剂(roche)的20mmtris-hcl(ph8.0),500mmnacl,3mmβ-巯基乙醇中的高压空化(10-15kpsi)将表达gst-il-37变体的细胞裂解。通过离心使细胞澄清,通过0.45μm膜过滤,并在4℃与谷胱甘肽sepharose4b树脂(gehealthcare)结合1小时。树脂用500ml20mmtris-hcl(ph8.0),200mmnacl和3mmβ-巯基乙醇洗涤。通过在4℃下用his-tev蛋白酶孵育过夜,从gst-标签释放蛋白质。蛋白质在20mmhepes(ph7.2),100mmnacl,2mmdtt和1mmedta中的hiloadsuperdex7516/60制备级柱(gehealthcare)上进一步纯化。
结晶和结构测定
在20℃通过悬滴蒸汽扩散在以1:1的液滴比例(dropratio)的2.1m硫酸铵和0.1m乙酸钠(ph4.5)中生长il-37晶体。在含有20%(v/v)甘油的母液中,将晶体在液氮中快速冷却。在澳大利亚同步加速器(australiansynchrotron)的mx2光束线(微聚焦)处以
尺寸排阻色谱和多角度光散射(sec-mals)
在10mmhepes(ph7.3),100mmnacl,1mmedta和2mmdtt中平衡的superdex7510/300柱(gehealthcare)上进行sec-mals测量。所有的实验均在25℃下在上述缓冲液中以0.4ml/min的流速进行。每次运行注入体积为110μl的6mg/ml的蛋白质。使用2mg/ml牛血清白蛋白(thermoscientificpierce)的测试注射用于校准目的。sec-mals系统包括shimadzudgu-20a5脱气机,lc-20ad液相色谱仪,sil-20aht自动进样器,cbm-20a通信总线模块,spd-20auv/vis检测器和cto-20ac柱箱,其偶联至安装有optilabt-rex折射率检测器的dawnheleo-ii多角度光散射检测器(wyatttechnology)。通过测量18个不同散射角处的散射光强度来计算摩尔质量。使用astra6.1软件(wyatttechnology)进行分子量计算。
细胞培养和转染。
pbmc实验经monashhealthhumanresearchethicscommitteeb批准,并在所有志愿者的明确书面同意下进行。如所述(15),通过密度梯度离心从健康志愿者的外周静脉血分离pbmc。将pbmc接种到含有1%v/v人血清和1:500mycozappr的rpmi培养基中,然后如所示用媒介物或recil-37预处理30分钟,然后用50pg/mllps或本文所述浓度的hklm、咪喹莫特、cpg-a或ssrna40处理20小时。然后将上清液进行细胞因子分析。thp-1细胞来源于atcc。它们总是在包含抗生素、抗真菌剂和抗支原体制剂的mycozapplus-cl(lonza)存在下培养。
如(16、1和5)中所述,用编码天然il-37b蛋白或单体d73k变体的构建体转染thp1细胞。简而言之,将每个il37b变体插入到具有gfp表达序列和组成型活性cmv启动子的pires载体中,并将il37b的c末端连接至flag。使用amaxanucleofectorkitv(thp1)和程序v001转染细胞,随后过夜恢复。转染后20小时,计数细胞并铺板。通过与50ng/mlpma孵育24小时,将转染的thp1细胞分化成巨噬细胞。之后,将培养基换成含有青霉素/链霉素和1%人血清的rpmi,并加入刺激物。在图例中指示的孵育时间段之后,收集上清液并在-80℃下储存直至分析。样品收集和分析以盲态方式进行。
elisa和多重elisa。
通过常规elisa(bd,elisakit.com)测量细胞因子。两种elisa方法均按制造商推荐的方法进行。
统计分析。
首先通过使用sigmaplot12.5(systatsoftwareinc.)测试数据集(原始数据)的常态性和同方差(p值至拒绝=0.05)。此后,应用适当的统计检验,包括非配对t检验(双尾检验,α=0.05),mann-whitney秩和检验,单因素方差分析(onewayanova)或单因素等级方差分析(onewayanovaonranks)。
动物实验。
涉及小鼠的所有程序均由莫纳什健康动物伦理委员会(monashhealthanimalethicscommittee)批准。c57bl/6野生型小鼠接受腹膜内注射40μg/kg的重组il-37变体或媒介物,接着60分钟后腹膜内注射lps(10mg/kg)。用il-37b转基因的纯合小鼠(1,5)也用lps(10mg/kg)或媒介物处理,用于直接比较。连续监测室温和湿度。如(1)中所述测量体温。在注射lps后24小时,麻醉小鼠并通过眼眶出血获得血浆,置于肝素化的管中。
试剂
人il-1β(beta)(elisa):目录号,557953,人il-6elisa,目录号:555220,鼠il-1β(beta)elisa,目录号:559603(bdbiosciences,newjersey,美国),大肠杆菌lps055:85,#l4005-100mg(sigma,stlouismo,美国),cpg-aodn2216innaxon#inax-200-005(adipogen,瑞士),咪喹莫特vaccigrade#vac-img,hklm#tlr1-hklm(invivogen,sandiego,ca,美国)。
实施例2
前体和成熟的il-37在溶液中形成同二聚体
细菌纯化的前体il-37(同种型b,在本文中称为il-37)的大小排阻色谱(sec)分析表明蛋白质在溶液中形成同二聚体(图1a和1b,左轴)。为了证实蛋白质是同二聚体,进行了与sec偶联的多角度光散射(mals)(图1b,右轴)。前体il-37(残基1-218)的测量分子量为43.9kda,与il-37二聚体的理论分子量48kda一致。这些数据与之前在人pbmc中检测到二聚体il-37(~45kda)和通过超速离心重组il-37(1,2)的研究相关。在图1b中,il-37(1-218)以9.8ml洗脱,il-37(21-218)以10ml洗脱,il-37(46-218)以11ml洗脱,il-18(37-193)以12.6ml洗脱。
为了研究从前体到成熟形式的n末端截短是否阻止了二聚化,从细菌细胞中纯化了il-37(残基21-218)和il-37(残基46-218)并进行了mals分析(图1a和1b)。成熟的il-37(21-218)和il-37(46-218)各自在溶液中形成二聚体,分子量分别为40.7kda和37.3kda。与il-37相比,成熟的il-18(残基37-193)具有与单体结构一致的17.3kda的测量分子量(图1b)(3)。
il-37同二聚体的晶体结构
为了解析il-37二聚化的分子基础,通过悬滴蒸汽扩散结晶成熟的il-37(残基46-218)。该结构通过分子置换解析并修正到
每个il-37亚单位由12个β股和三个α螺旋构成,其形成il-1超家族的特征性β-三叶形折叠(图1c)。四条β股的三条假重复(pseudorepeat)包装在一起,每条重复的两股形成一个六股β-桶,其余两股形成一个六股封端区域(cappingregion)。反平行β-桶中的n末端和c末端股β1和β12的相互作用关闭了β-三叶形折叠。三个α-螺旋装饰β-三叶形折叠的外侧,螺旋α1和α2位于股β7和β8之间,且位于股β11之后有一个短的310螺旋。
表3.数据收集和修正统计
*括号中的值是最高分辨率的壳,数据集来自单晶。
il-37同二聚体具有头对头对称和高度有组织的界面
对称的头对头il-37二聚体界面总共埋有
为了验证界面的分子细节并产生单体il-37,设计了许多靶向干扰il-37二聚化的结构指导的突变。asp73在界面内与lys83形成离子相互作用,asp73上的切换电荷突变(d73k)有效地消除了界面,其具有与il-37单体对应的18.8kda的分子量(图2d)。tyr85位于疏水核心处,并且向界面贡献大的掩埋表面区域,并且突变为丙氨酸(y85a)也破坏二聚体界面,其具有通过sec-mals测量的18.2kda的分子量(图2d;il-37wt以11ml洗脱,il-37d73a以11.6ml洗脱,il-37d73k以12.7ml洗脱,il-37y85a以12.6ml洗脱)。归因于il-37二聚体界面的对称性,每个点突变都有效地靶向界面中的两个相互作用位点,并可能进一步辅助二聚体界面的破坏。
为了评估单体-二聚体界面中的改变,目前已经通过sec-mals测试了五种il-37突变体(图3;il-37d73a+k83a以11.1ml洗脱,il-37k83e以11.6ml洗脱,针对其他突变体和野生型的洗脱参考上面与图2d有关的内容)位于界面的核心并且突变成丙氨酸(y85a)将il-37转变为单体。asp73位于界面的边界处并与lys83形成离子相互作用。asp73突变成丙氨酸(d73a)部分破坏了二聚体界面,使平衡向单体转移。推测asp73的电荷转换突变(d73k)通过与lys83的电荷排斥来消除二聚体界面。lys83的电荷转换突变(k83e)使平衡向单体转移,但在消除界面方面不如d73k有效。有趣的是,完全去除门控(gating)界面的离子相互作用(d73a/k83a)对平衡似乎只有很小的影响。这些突变表明离子门控相互作用不是界面形成所必需的。相反,β3-β4环区域的埋藏的tyr85侧链和氢键可能更关键。
实施例3
两种方法被用来评估il-37单体化的功能影响。首先,用重组(rec)il-37的不同变体处理新鲜的人pbmc,所述重组(rec)il-37包括甚至在非常低的浓度下自发二聚化的天然蛋白质(野生型),以及其中二聚化被氨基酸73的d至k的突变所阻止的变体。这两种变体在氨基酸21和46被n末端截短进行测试。
正如以前的研究所预期的那样,natil-37(野生型recil-37)赋予lps诱导的il-1β蛋白的适度降低(natil-37(21-218)多达40%,natil-37(46-218)多达52%;图5,左边组)。有趣的是,将natil-37的浓度提高到超过10ng/ml,例如100ng/ml,大大降低了natil-37的有效性(图5),实际上,在三个供体中,100ng/mlnatil-37(46-218)诱导促炎反应(il-1β增加多达36%)。
il-37单体化显著增加这些抗炎活性。在所有测试浓度下,monoil-37(d73k)在阻断lps处理的pbmc中的il-1β释放比natil-37更有效(图5)。在10pg/ml时,monoil-37b(46-218)使il-1β降低59%,比natil-37(46-218)提高近2倍,并且在100ng/mlmonoil-37(46-218)比natil-37b(46-218)更有效超过5倍。此外,monoil-37b在较高浓度下保持其抗炎活性;因此,二聚体形成可能在这样的浓度有助于降低il-37的活性,或者实际上将其转化为促炎症活性,如100ng/ml的21-218变体的情况(monoil-37b降低了21%的il-1β,相比之下,natil-37增加了26%的il-1β)。对于monoil-37(46-218),浓度范围进一步扩大,在1pg/ml(il-1β降低了52%)和1μg/ml观察到抗炎作用。此外,抗炎功能的丧失发生在比natil-37高10倍的浓度下(1μg/ml)。值得注意的是,在浓度范围的1pg/ml低端,单体46-218recil-37b仍然是10pg/ml天然二聚体recil-37活性的1.6倍(图5)。n末端截短也影响il-37功能:通过比较二者、天然以及d73k突变体与它们的对应物(图5,组1相对于组3和组2相对于组4;从左到右指定为组1到组4),46-218变体在阻断il-1β产生方面比21-218变体有效得多。
为了考虑il-37的细胞内作用机制,通过转染入thp-1巨噬细胞评估了il-37单聚化对其抗炎效力的影响。因此,第一次比较了thp-1巨噬细胞中二聚体天然il-37b(natil-37)与突变单体il-37b(d73k,monoil-37)的抗炎活性(图6)。与对照转染相比,转染全长natil-37减少了71%的lps诱导的il-1β蛋白丰度(271至80pg/ml)。monoil-37的抗炎活性强2.7倍,减少89%的il-1β(271至30pg/ml)。与natil-37不同,monoil-37也减少媒介物处理的培养物中的il-1β。这些数据表明monoil-37的抗炎活性大于二聚体natil-37的抗炎活性,il-37生物活性不需要二聚化。
还在pbmc和thp-1培养物的上清液中评估了tnf的蛋白质丰度,揭示了il-37也阻断了这种促炎细胞因子,尽管其效力略低于il-1β(数据未显示)。
实施例4
内毒素休克的体内模型
为了增加这些发现的体内维度,使用了内毒素休克的小鼠模型。c57bl/6野生型小鼠接受腹膜内注射重组il-37变体(40μg/kg)或媒介物,接着腹膜内注射lps。il-37转基因小鼠也用lps或媒介物处理,用于直接比较。尽管每个il-37变体都能改善内毒素休克(减少低温和血浆il-1β,图7和8),两种单体il-37蛋白的抗炎活性高于其二聚体natil-37对应物的抗炎活性。值得注意的是,monoil-37(46-218)提供的保护几乎与il-37转基因赋予的保护一样强。事实上,monoil-37(46-218)组和未接受lps的小鼠体温和血浆il-1β之间的差异最小。
实施例5
在位置85从酪氨酸到丙氨酸的突变(除了d73k突变之外)是破坏il-37二聚体界面的另一途径。每个亚单位的位置85处的部分疏水性酪氨酸残基被掩埋在由二聚体界面形成的疏水口袋中。将该残基突变为丙氨酸显著减少了这些相互作用并破坏了二聚体界面。因此,recil-37y85a不能形成同二聚体。与单体il-37比二聚体il-37b具有更高的生物学活性的概念一致,与recil-37d73k相比,特别是与天然recil-37b相比,在用tlr2激动剂热灭活的单核细胞增生李斯特氏菌体外刺激的pbmc中(hklm,图9)和用tlr4激动剂lps体内注射的小鼠中,recil37y85a的抗炎作用增加(图7)。然而,在分别用tlr7和tlr9的配体,咪喹莫特和cpg-a刺激的pbmc中,recil-37d73k和y85a之间的生物活性几乎没有差异。
总体而言,y85a变体的数据支持这样的概念,即当用作细胞外处理时,将il-37单体至二聚体平衡移向单体增加了il-37的抗炎特性。
实施例6
recil-37和单体变体对lps以外的tlr激动剂诱导的炎症的作用
il-37是由各种炎症攻击引发的炎症的强大缓冲剂。在体外,本发明人已经显示,除了lps外,这些攻击还包括tlr1激动剂pam3csk4(nold等人,natimmunol2010中的图1),il-1β(nold等人,natimmunol2010中的图3和4),lps+il-12、tnf和il-12+il-18(nold等人,natimmunol2010中的图7和nold-petry等人,natimmunol2015中的图4)。此外,il-37在大量动物疾病模型中发挥保护作用;除内毒素休克模型外,这些包括dss结肠炎、斑色鱼鳞癣、接触性超敏反应和其他疾病。
这种针对广泛的炎症触发因子的活性具有高度的转化相关性(translationalrelevance),因为因此可以预料,il-37将有效改善同样广泛的疾病中的炎症,从普通感冒到牛皮癣、心肌梗塞、中风和许多其他疾病。
然而,通过使用转染来表达il-37的细胞或转il-37基因的小鼠品系产生nold等人,natimmunol2010和nold-petry等人,natimmunol2015中所述的数据。由于il-37具有双重作用机制,包括细胞内和细胞外信号传导途径,重要的是建立了可能几乎专门通过细胞外il-37受体介导的途径起作用的recil-37的处理不但有效地阻断由lps引发的炎症,而且还阻断由其他炎症剂引发的炎症。本文描述的数据表明情况确实如此。
在用tlr2激动剂hklm刺激的人pbmc中,recil-37赋予il-1β降低多达51%,并使il-6蛋白丰度降低多达36%(图9)。在用下述处理的培养物中,促炎细胞因子的丰度也更低:tlr7配体咪喹莫特+recil-37b(减少多达36%的il-1β,图10),tlr9激动剂cpg-a+recil-37b(减少多达30%的il-6,图11)和tlr8的配体ssrna40(减少多达49%的il-1β,图12)。
此外,本文的数据强烈表明,单体recil-37的抗炎作用大于能够形成同二聚体的recil-37的抗炎作用。图5显示,在用tlr4配体lps刺激的pbmc中,单体d73k变体在大多数浓度下比其天然二聚对应物显著更有活性。在用tlr2和7、8和9的配体刺激的pbmc中,也有类似的观察结果:y85a变体在阻断hklm(tlr2配体)和咪喹莫特(tlr7配体)诱导的il-1β或il-6中比天然recil-37显著更有活性(分别为图9和10)。此外,在咪喹莫特和cpg-a(tlr9配体)刺激的培养物中,由d73k和y85a赋予的对il-1β和il-6的抑制是显著的,而由natil-37b赋予的则不是(分别见图10和11)。尽管对于tlr8的配体ssrna40统计学分析是不可能的,因为只有两个供体被研究,d73k在测试的每种浓度下均比natil-37在阻断il-1β中显著更有效(图12)。
il-37的单体变体在临床上会是在广泛的炎性损伤引起的各种疾病中阻断炎症的有效的药物活性成分。
参考文献
1.nold,m.f.,nold-petry,c.a.,zepp,j.a.,palmer,b.e.,bufler,p.,anddinarello,c.a.(2010)il-37isafundamentalinhibitorofinnateimmunity.nat.immunol.11,1014–1022
2.kumar,s.,hanning,c.r.,brigham-burke,m.r.,rieman,d.j.,lehr,r.,khandekar,s.,kirkpatrick,r.b.,scott,g.f.,lee,j.c.,lynch,f.j.,gao,w.,gambotto,a.,andlotze,m.t.(2002)interleukin-1f7b(il-1h4/il-1f7)isprocessedbycaspase-1andmatureil-1f7bbindstotheil-18receptorbutdoesnotinduceifn-gammaproduction.cytokine.18,61–71
3.kato,z.,jee,j.,shikano,h.,mishima,m.,ohki,i.,ohnishi,h.,li,a.,hashimoto,k.,matsukuma,e.,omoya,k.,yamamoto,y.,yoneda,t.,hara,t.,kondo,n.,andshirakawa,m.(2003)thestructureandbindingmodeofinterleukin-18.nat.struct.biol.10,966–971
4.tsutsumi,n.,kimura,t.,arita,k.,ariyoshi,m.,ohnishi,h.,yamamoto,t.,zuo,x.,maenaka,k.,park,e.y.,kondo,n.,shirakawa,m.,tochio,h.,andkato,z.(2014)thestructuralbasisforreceptorrecognitionofhumaninterleukin-18.natcommun.5,5340
5.nold-petry,c.a.,lo,c.y.,rudloff,i.,elgass,k.d.,li,s.,gantier,m.p.,lotz-havla,a.s.,gersting,s.w.,cho,s.x.,lao,j.c.,ellisdon,a.m.,rotter,b.,azam,t.,mangan,n.e.,rossello,f.j.,whisstock,j.c.,bufler,p.,garlanda,c.,mantovani,a.,dinarello,c.a.,andnold,m.f.(2015)il-37requiresthereceptorsil-18rαandil-1r8(sigirr)tocarryoutitsmultifacetedanti-inflammatoryprogramuponinnatesignaltransduction.nat.immunol.16,354–365(2015).
6.krissinel,e.,andhenrick,k.(2007)inferenceofmacromolecularassembliesfromcrystallinestate.j.mol.biol.372,774–797
7.matsuura,y.,andstewart,m.(2004)structuralbasisfortheassemblyofanuclearexportcomplex.nature.432,872–877
8.collaborativecomputationalproject,number4(1994)theccp4suite:programsforproteincrystallography.actacrystallogr.dbiol.crystallogr.50,760–763
9.dunn,e.f.,gay,n.j.,bristow,a.f.,gearing,d.p.,o'neill,l.a.j.,andpei,x.y.(2003)high-resolutionstructureofmurineinterleukin1homologueil-1f5revealsuniqueloopconformationsforreceptorbindingspecificity.biochemistry.42,10938–10944
10.bunkóczi,g.,echols,n.,mccoy,a.j.,oeffner,r.d.,adams,p.d.,andread,r.j.(2013)phaser.mrage:automatedmolecularreplacement.actacrystallogr.dbiol.crystallogr.69,2276–2286
11.adams,p.d.,afonine,p.v.,bunkóczi,g.,chen,v.b.,davis,i.w.,echols,n.,headd,j.j.,hung,l.-w.,kapral,g.j.,grosse-kunstleve,r.w.,mccoy,a.j.,moriarty,n.w.,oeffner,r.,read,r.j.,richardson,d.c.,richardson,j.s.,terwilliger,t.c.,andzwart,p.h.(2010)phenix:acomprehensivepython-basedsystemformacromolecularstructuresolution.actacrystallogr.dbiol.crystallogr.66,213–221
12.busterversion2.10.0
13.emsley,p.,andcowtan,k.(2004)coot:model-buildingtoolsformoleculargraphics.actacrystallogr.dbiol.crystallogr.60,2126–2132
14.chen,v.b.,arendall,w.b.,headd,j.j.,keedy,d.a.,immormino,r.m.,kapral,g.j.,murray,l.w.,richardson,j.s.,andrichardson,d.c.(2010)molprobity:all-atomstructurevalidationformacromolecularcrystallography.actacrystallogr.dbiol.crystallogr.66,12–21
15.noldm.,etal.il-18bpa:fccooperateswithimmunosuppressivedrugsinhumanwholeblood.biochempharmacol66,505-510(2003).
16.noldm.f.,etal.endogenousil-32controlscytokineandhiv-1production.jimmunol181,557-565(2008).
序列表
<110>莫纳什大学(monashuniversity)
亨利王子医学研究所(princehenry'sinstituteofmedicalresearch)
<120>il-37变体
<130>m50134783
<150>2015902262
<151>2015-06-15
<150>2016900703
<151>2016-02-26
<160>1
<170>siposequencelisting1.0
<210>1
<211>218
<212>prt
<213>智人(homosapiens)
<400>1
metserphevalglygluasnserglyvallysmetglysergluasp
151015
trpglulysaspgluproglncyscysleugluaspproalaglyser
202530
proleugluproglyproserleuprothrmetasnphevalhisthr
354045
serprolysvallysasnleuasnprolyslyspheserilehisasp
505560
glnasphislysvalleuvalleuaspserglyasnleuilealaval
65707580
proasplysasntyrileargprogluilephephealaleualaser
859095
serleuserseralaseralaglulysglyserproileleuleugly
100105110
valserlysglygluphecysleutyrcysasplysasplysglygln
115120125
serhisproserleuglnleulyslysglulysleumetlysleuala
130135140
alaglnlysgluseralaargargpropheilephetyrargalagln
145150155160
valglysertrpasnmetleugluseralaalahisproglytrpphe
165170175
ilecysthrsercysasncysasngluprovalglyvalthrasplys
180185190
phegluasnarglyshisileglupheserpheglnprovalcyslys
195200205
alaglumetserprosergluvalserasp
210215
- 还没有人留言评论。精彩留言会获得点赞!