S1PR2拮抗剂及其用途的制作方法
该技术通常涉及方法,其通过s1pr2抑制用于治疗遗传性致盲性障碍家族性渗出性玻璃体视网膜病变(fevr)。该技术还涉及化合物,其含有jte-013脲基的生物电子等排体替代物及其类似物,及它们在治疗视网膜病变和特征在于血管生成不足的疾病中的用途。
背景技术:
视网膜是衬在眼睛后面的神经组织薄层,负责感知视觉刺激。在发育过程中,视网膜脉管系统由内皮出芽(sprouts)开始,所述内皮出芽位于主动脉和静脉上,所述主动脉和静脉从视神经盘放射状向外伸出至视网膜周边,具有一对位于神经元中央层两侧的毛细血管床进一步穿透视网膜。一旦到位,主脉管系统经历成熟以明确动脉和静脉,新生的网络被修剪,并形成血-视网膜屏障。血管平滑肌细胞和周细胞(也称为壁细胞,包裹毛细血管内皮细胞的收缩细胞)的募集有助于稳定新形成的血管。控制血管在眼中迁移和图式形成(patterning)的分子机制尚不清楚。
在人类中,视网膜血管发育通常在约足月分娩时完成,但在fevr中会被延迟或被阻滞。fevr的特征在于由于周边视网膜血管化失败导致视网膜血管化不足(hypovascularization),接着继发性异常新血管形成。严重形式的fevr存在双侧先天性视网膜皱褶(retinalfolds)或视网膜脱离(图1)。目前,fevr的处理是通过激光和手术。虽然干预提高了保持视力的机会,尽管目前做出了最大的努力,超过75%的眼睛仍然法定失明。尽管有当前的激光和手术治疗,视力丧失仍发生在大多数的fevr患者中。这种病症的理想治疗是使早期检测伴随早期介入治疗,这将完全防止并发症的进展。
概要
公开的附加特征和优点将在以下说明书中描述,说明书中部分内容是显而易见的,或者可以通过实施本公开的原理来习得。本公开的特征和优点可以通过所附权利要求中特别指出的手段和组合来实现和获得。从以下说明书和所附权利要求,本公开的这些和其他特征将变得更加显而易见,或者可以通过实施本文阐述的原理而习得。
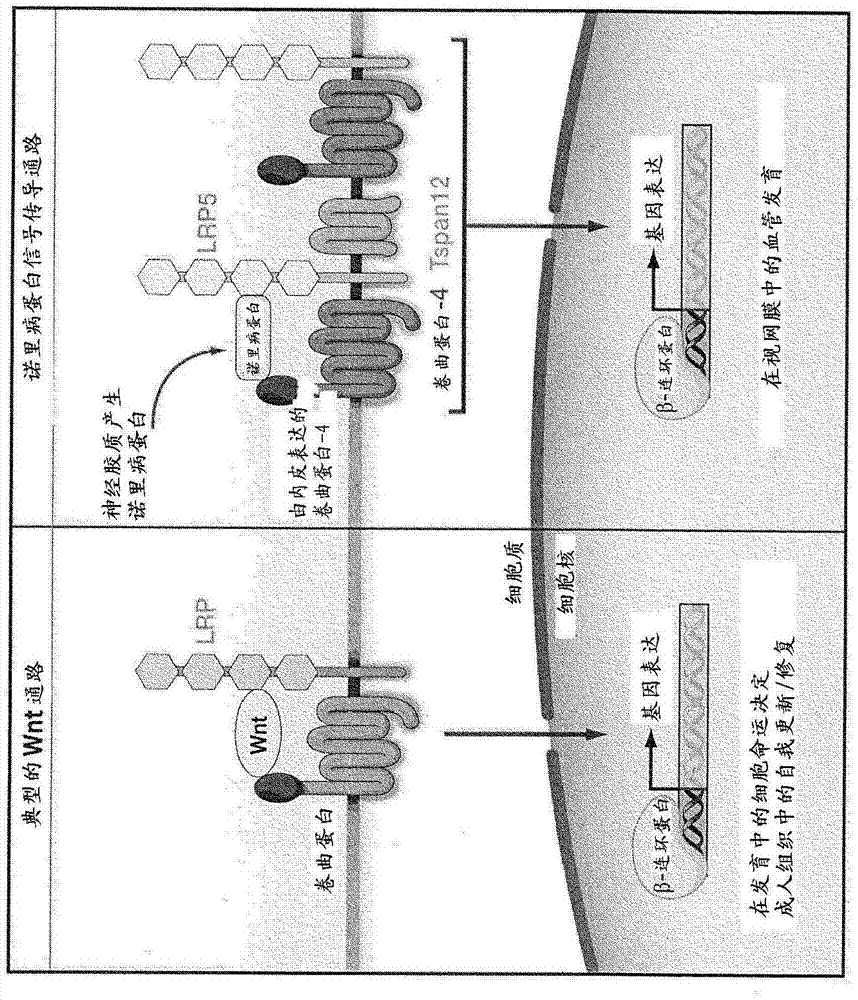
对当突变时引起fevr的基因的发现,增加了对调节视网膜血管发育的分子通路的了解。到目前为止,已经鉴定了导致fevr的五种基因:fzd4,lrp5,tspan12,ndp和znf408。已知导致fevr的五种基因中的四种形成卷曲蛋白(frizzled)受体信号传导复合物(图2)。fzd4是七种跨膜受体的卷曲蛋白家族的一部分,所述跨膜受体通常由wnt配体家族激活。fzd4在卷曲蛋白受体家族中是独特的,因为它是由非wnt配体诺里病蛋白(norrin)(ndp基因的产物)特异性激活的。诺里病蛋白(norrin)由müller胶质细胞分泌,并与位于血管内皮细胞上的fzd4受体结合。lrp5是fzd4的共受体,并且是fzd4起作用所必需的。在fzd4、ndp和lrp5中的突变已经显示出引起了fevr。(mutantfrizzled-4disruptsretinalangiogenesisinfamilialexudativevitreoretinopathy.robitaillej,macdonaldml,kaykasa,sheldahllc,zeislerj,dubémp,zhanglh,singarajarr,guernseydl,zhengb,siebertlf,hoskin-motta,tresemt,pimstonesn,shastrybs,moonrt,haydenmr,goldbergyp,samuelsme.naturegenetics,32,326-30(2002))(autosomalrecessivefamilialexudativevitreoretinopathyisassociatedwithmutationsinlrp5.xiaodongjiao,valerioventruto,michaelt.trese,barkurs.shastry,j.fieldinghejtmancik.amjhumgenet75,p878–884(2004))(amutationinthenorriediseasegene(ndp)associatedwithx-linkedfamilialexudativevitreoretinopathy.z-y.chen,e.m.battinelli,a.fielder,s.bundey,k.sims,x.o.breakefield&i.w.craig.naturegenetics5,180-183(1993))。
tspan12在内皮细胞中表达,直接与fzd4结合,并增强了诺里病蛋白、fzd4和lrp5之间的相互作用。不清楚fzd4是通过典型的wnt通路(图2)发出信号,还是通过非典型的wnt通路发出信号。已经显示出tspan12中的突变引起fevr。(recessivemutationsintspan12causeretinaldysplasiaandseverefamilialexudativevitreoretinopathy(fevr).jamesa.poulter;alicee.davidson;manirali;davidf.gilmour;davida.parry;helena.mintz-hittner;ianm.carr;helenm.bottomley;vernonw.long;louisem.downey;panagiotisi.sergouniotis;genevievea.wright;roberte.maclaren;anthonyt.moore;andrewr.webster;chrisf.inglehearn;carmeltoomes.iovs53,2873-2879(2012).)(mutationsintspan12causeautosomal-dominantfamilialexudativevitreoretinopathy.poulterja1,alim,gilmourdf,ricea,kondoh,hayashik,mackeyda,kearnsls,ruddlejb,craigje,pierceea,downeylm,mohamedmd,markhamaf,inglehearncf,toomesc.amjhumgenet86,248-53(2010))。
对fzd4,tspan12,lrp5和ndp的小鼠敲除模型充当在fevr患者中观察到的眼部表型的准确模拟物。这些模型已经允许用于fevr表型的详细分析。来自小鼠研究的一个重要观察结果是,虽然视网膜脉管系统在fevr的小鼠模型中受损,但是视网膜本身显示出在形态上正常,提供了可以防止视力丧失的用于干预的机会窗。
1-磷酸鞘氨醇(sphingosine-1-phosphate)(s1p)是血液携带的脂质第二信使,其通过鞘磷脂酶、神经酰胺酶和鞘氨醇激酶的作用从鞘磷脂代谢产生(图3)。s1p生成的主要部位是内皮细胞和红细胞。s1p激活g蛋白偶联受体的内皮分化家族,命名为s1pr1-5(以前称为edg1-5)。s1pr在不同细胞类型中表达,并调节许多生物学过程。s1pr1,-2和-3功能特别令人感兴趣,因为它们在血管内皮细胞上表达并调节血管发育和稳定性。(sphingosine1-phosphatereceptorsignaling.rosenh,gonzalez-cabrerapj,sannamg,browns.annurevbiochem.78,743-68(2009).)(sphingosine1-phosphateandcancer.pyne,nj,pyne,s.natrevcancer10,489-503(2010))。
s1pr1对于血管稳定化至关重要,并且增加血管迁移。s1pr1与gi偶联并激活磷脂酰肌醇3-激酶通路,其通过rac影响肌动蛋白组装和细胞迁移。已经报道了对于s1pr3与gq偶联的类似重叠功能。相反的,s1pr2拮抗s1pr1和-3信号传导。s1pr2主要激活g12/13并激活rho-rho激酶通路并抑制rac功能(图4)。这些拮抗s1pr通路之间的平衡决定了内皮细胞对s1p的响应。(sphingosine1-phosphatereceptorsignaling.rosenh1,gonzalez-cabrerapj,sannamg,browns.annurevbiochem.78,743-68(2009).)(sphingosine1-phosphatereceptorsignaling.rosenh1,gonzalez-cabrerapj,sannamg,browns.annurevbiochem.78,743-68(2009).)。
本技术提供了组合物和方法,其通过施用治疗有效量的s1pr2抑制剂来安全且有效地治疗患有fevr的个体。
在进一步的方面,该技术提供了试剂盒,其包含含有s1pr2抑制剂的药物组合物,其可以包括小分子或生物制品,以及用于向受试者施用用于治疗患有fevr的受试者的组合物的说明书。
如本文所使用的,术语“抑制”是指蛋白质生物学活性的降低,优选为人蛋白质s1pr2活性的降低。
如本文所使用的,术语“基因”是指核酸分子,其编码特定蛋白质或在某些情况下为功能性或结构性rna分子。
如本文所使用的,“蛋白质”和“多肽”同义地用于指任何肽连接的氨基酸链,而不考虑长度或翻译后修饰,例如糖基化或磷酸化。
当提及核酸分子或多肽时,术语“野生型”是指天然存在的(例如天然的,wt)核酸或多肽。
如本文所使用的,术语“治疗”和“疗法”定义为将治疗剂应用或施用于患者或受试者,或将治疗剂应用或施用于来自患者或受试者的分离的组织或细胞系,所述患者或受试者具有障碍或疾病、障碍或疾病的症状、或对障碍或疾病的倾向,其目的是消除,治愈,减轻,缓解,改变,补救,改善,提高或影响障碍或疾病、障碍或疾病的症状、或对障碍或疾病的倾向。
如本文所使用的,术语“治疗有效量”是指将引起期望的治疗效果或响应的s1pr2抑制剂的量。
术语“患者”,“受试者”和“个体”在本文中可互换使用,并且是指待治疗、诊断和/或从中获得生物样品的哺乳动物(例如人,啮齿动物,非人灵长类动物,犬科动物,牛科动物,绵羊科动物,马科动物,猫科动物等)受试者。
如本文所用的术语“试剂盒”是指包含组分的包装产品,使用它施用治疗有效量的s1pr2抑制剂来治疗fevr。试剂盒优选包含容纳试剂盒组分的盒子或容器。所述盒子或容器贴有标签或食品药品管理局批准的方案。盒子或容器所容纳的技术组分优选地盛放在塑料,聚乙烯,聚丙烯,乙烯或丙烯器皿内。这些器皿可以是加盖的管子或瓶子。该试剂盒还可以包括用于施用s1pr2抑制剂的说明书。
本发明部分源自以下发现:s1pr2活性的抑制(例如,通过施用s1pr2拮抗剂)能够改善受试者的视网膜血管化,否则将在视网膜发育期间呈现血管化不足(hypovascularization)或驱血(avascularization),接着异常眼新血管形成,其可损害视网膜完整性和功能,例如,在患有fevr的受试者中。具体而言,如本文所示,s1pr2拮抗剂的施用可改善在该发育性疾病中发生的发育性血管化不足或驱血,并且允许在视网膜发育期间建立血管床,从而避免有害的异常新血管形成,其通常会在这样的患病受试者中出现,s1pr2拮抗剂的总体效果是改善视网膜血管化。
此外,本发明涉及通式(ix)的s1pr2拮抗剂,其中z不是通式(x)的–(c=o)–,如下所述,其特别是用于治疗视网膜病变或特征在于血管生成不足的疾病。s1pr2拮抗剂可以施用于有其需要的受试者,并且可以包含在本文所述的药物组合物中。视网膜病变可以是糖尿病性视网膜病变,黄斑变性,高血压性视网膜病变,放射性视网膜病变,日光性视网膜病变,早产儿视网膜病变(rop),诺里病(nd),家族性渗出性玻璃体视网膜病变(fevr),coats病,镰状红细胞性视网膜病变,色素性视网膜炎等。特征在于血管生成不足的疾病可以是动脉粥样硬化,高血压,糖尿病,再狭窄,先兆子痫,月经过多,新生儿呼吸窘迫,肺纤维化,肾病,骨质疏松症,肌萎缩性侧索硬化,中风,阿尔茨海默氏病等。
在本文所述的s1pr2拮抗剂的一个方面中,本发明提供了一种在处于视网膜血管化不足或驱血的风险的受试者中诱导建立正常视网膜血管床的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的fzd4活性的受试者中使视网膜血管化正常化的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的fzd4活性的受试者中诱导建立正常视网膜血管床的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的ndp活性的受试者中使视网膜血管化正常化的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的ndp活性的受试者中诱导建立正常视网膜血管床的方法,所述方法包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的tspan12活性的受试者中使视网膜血管化正常化的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的tspan12活性的受试者中诱导建立正常视网膜血管床的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的lrp5活性的受试者中使视网膜血管化正常化的方法,包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供用于在视网膜脉管系统中具有降低的lrp5活性的受试者中诱导建立正常视网膜血管床的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中诱导视网膜正常血管化的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中增加视网膜血管化的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视网膜血管化不足或驱血的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中诱导建立正常视网膜血管床的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供用于在患有或处于fevr的风险中的受试者中减少周边视网膜血管化不足或驱血的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在本文所述的一些实施方式中,可以在异常低血管性或无血管性视网膜区域建立/出现之前施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中诱导周边视网膜的正常血管化的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中增加周边视网膜血管化的方法,其包括向受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中抑制视网膜的异常新血管形成的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视网膜完整性丧失的方法,包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中的减少视力丧失的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视网膜脱离的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
另外,在一个方面,本发明提供了用于使处于视网膜血管化不足或驱血的风险中的受试者的视网膜血管化正常化的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了一种组合物,其用于在处于视网膜血管化不足或驱血的风险中的受试者中诱导建立正常视网膜血管床。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜脉管系统中具有fzd4活性降低的受试者中使视网膜血管化正常化的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供一种组合物,其用于在视网膜脉管系统中具有降低的fzd4活性的受试者中诱导正常视网膜血管床的建立。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的ndp活性的受试者中使视网膜血管化正常化的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的ndp活性的受试者中诱导建立正常视网膜血管床的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的lrp5活性的受试者中使视网膜血管化正常化的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的lrp5活性的受试者中诱导建立正常视网膜血管床的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜脉管系统中具有降低的tspan12活性的受试者中使视网膜血管化正常化的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在视网膜血管中具有tspan12活性降低的受试者中诱导建立正常视网膜血管床的组合物。在一个实施方式中,受试者处于随之发生的异常新血管形成的风险中。在一个实施方式中,受试者处于随之发生的视网膜完整性丧失或视网膜脱离的风险中。在一个实施方式中,受试者患有或处于fevr的风险中。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中诱导视网膜的正常血管化的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中增加视网膜血管化的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视网膜血管化不足或驱血的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者的视网膜中建立正常视网膜血管床的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少周边视网膜血管化不足的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中诱导周边视网膜的正常血管化的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中增加周边视网膜血管化的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供用于在患有或处于fevr的风险中的受试者中抑制视网膜的异常新血管形成的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视网膜完整性丧失的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视力丧失的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了用于在患有或处于fevr的风险中的受试者中减少视网膜脱离的组合物。
在一个实施方式中,组合物是包含s1pr2拮抗剂的药物组合物,该药物组合物用于s1pr2拮抗剂的玻璃体内施用。
在一个方面,本发明提供了治疗患有或处于fevr的风险中的受试者的方法,其包括向所述受试者施用治疗有效量的s1pr2拮抗剂。
在一个方面,本发明提供用于治疗fevr的药物组合物,其包含治疗有效量的s1pr2拮抗剂。
在一个实施方式中,药物组合物用于s1pr2拮抗剂的玻璃体内递送。
在进一步的方面,本发明提供了包含本发明药物组合物的试剂盒,其可包括说明书,其用于将所述药物组合物施用给有其需要的受试者。在一个实施方式中,该试剂盒用于治疗fevr。在一个实施方式中,所述试剂盒用于在处于视网膜血管化不足或驱血的风险中的受试者中诱导建立正常视网膜血管床的治疗,所述方法包括向所述受试者施用治疗有效量的s1pr2拮抗剂。在一个实施方式中,所述试剂盒用于治疗随之而来的异常新血管形成。在一个实施方式中,所述试剂盒用于治疗随之而来的视网膜完整性丧失或视网膜脱离。
本公开的附加特征和优点将在以下说明书和所附权利要求中阐述。
附图说明
为了描述上述记载的方式和通过本公开可以获得的其他优点以及特征,通过参考附图中所示的具体实施方式,将对上述简要描述的原理进行更具体的描述。应当理解,这些附图仅描绘了本公开的示例性实施方式,并且因此不被认为是对其范围的限制,通过使用附图以其他特征和细节来描述和解释本文的原理,其中:
图1示出了人眼的血管化。(a)通常,视网膜脉管系统从视神经盘向外伸出到视网膜周边。家族性渗出性玻璃体视网膜病变(fevr)是一种遗传性障碍,其导致视网膜血管化不足(b),并接着异常新血管形成,可导致视网膜脱离(c)。
图2示出了与诺里病蛋白通路相比的典型wnt通路。通过卷曲蛋白-4(frizzled-4)受体的诺里病蛋白信号传导调节视网膜中的血管发育,其被假设为通过β-连环蛋白发出信号。诺里病蛋白,卷曲蛋白-4,lrp5和tspan12的基因编码中的突变引起家族性渗出性玻璃体视网膜病变(fevr)。参见cell139,227-29(2009)。
图3示出了1-磷酸鞘氨醇(sphingosine-1-phosphate)(s1p)的产生通路。
图4示出了1-磷酸鞘氨醇受体(s1pr)信号传导通路。在血管内皮细胞上发现s1pr1、2和3。s1pr1和s1pr3驱动血管迁移,而s1pr2抵消它们的作用。在该程度上,抑制s1pr2(抑制血管迁移抑制剂)将产生对于视网膜血管发育障碍家族性渗出性玻璃体视网膜病变(fevr)的正常视网膜血管化的恢复。(图片来自nat.rev.cancer10,489-503(2010))。
图5示出了在s1pr2基因失活时,fevr的tspan12-/-小鼠模型中脉管系统图式形成(patterning)的改善。将视网膜平放在p17处,并在用异凝集素b4alexafluor594染色后通过共聚焦显微镜可视化脉管系统。
图6示出了在s1pr2基因失活时,fevr的fzd4-/-小鼠模型中脉管系统图式形成的改善。将视网膜平放在p17处,并在用异凝集素b4alexafluor594染色后通过共聚焦显微镜可视化脉管系统。
图7示出了用s1pr2拮抗剂jte-013处理fzd4-/-小鼠改善了fevr表型。从p7到p25每隔一天给fzd4-/-小鼠腹膜内注射0.5mg/kgjte-013。将视网膜平放在p25处,并且在用异凝集素b4alexafluor594染色后通过共聚焦显微镜可视化脉管系统。在用jte-013处理的小鼠中正常的视网膜脉管系统图式形成基本恢复。
图8示出了1-磷酸鞘氨醇受体2(s1pr2)结合口袋。识别出位于蛋白质细胞外面上(extracellularface)由34个氨基酸组成的口袋,并且与其对应的1-磷酸鞘氨醇受体相关。氨基酸如下:tyr18,lys22,glu23,leu25,glu26,gln28,glu29,thr30,arg33,ala36,ser37,ile40,phe86,asn89,thr90,leu92,ser93,gly94,ser95,thr97,leu98,trp105,arg108,glu109,val182,leu183,pro184,tyr268,lys269,ala270,his271,tyr272,phe274和ala275。这些氨基酸位于蛋白质的跨膜结构域i,ii,iii和vii上。用jte-013结合显示人s1pr2配体结合口袋。jte-013是一种对s1pr2具有18nm亲和力的化合物,并且对s1pr2比对其他s1pr的特异性高100倍。使用s1pr2结合口袋的分子建模来鉴定s1pr2拮抗剂。
图9示出了1-磷酸鞘氨醇及其经鉴定的靶区域。这些经鉴定的化合物与结合口袋相互作用比其与已知受体相互作用更强烈,从而阻止s1p结合。经鉴定的化合物商售可得。
图10示出了成年斑马鱼中fzd4基因的失活导致在人和小鼠中观察到的异常新血管形成。为了靶向定位fzd4基因,创建了一对talen核酸酶。使携带相当大比例突变的创始者与fli1:egfp转基因鱼(用于脉管系统的gfp标记物)交配,并且所得的f1鱼生长至成年。在fzd4基因的开放阅读框中验证了10个核苷酸的插入。使成对的fzd4杂合子鱼交配以产生含有纯合子突变体的后代。纯合子突变型鱼没有任何明显的胚胎表型,并长到成年。为了检查视网膜脉管系统是否受到fzd4突变的影响,从野生型和突变型鱼中解剖出视网膜并平放,并通过共焦显微镜可视化。纯合子fzd4-/-突变型大面积无血管且在血管化的区域中血管形态异常。
图11a-d示出了经鉴定为有活力的新型s1pr2拮抗剂的21种化合物。
发明详述
下面详细讨论本公开的各种实施方式。虽然讨论了具体的实施方式,但应该理解为这仅仅是为了说明目的而完成的。在本公开内容中引用的所有参考文献并入本文。相关领域的技术人员会意识到,在不脱离本公开的精神和范围的情况下可以使用其他组分和构型。
描述了通过施用治疗有效量的s1pr2拮抗剂来治疗视网膜血管障碍的组合物和方法。在优选的实施方式中,治疗方案适合于治疗fevr。
s1pr2拮抗剂可以是由以下通式(ix)表征的化合物:
其中
r1是c1-c12烷基;
r2,r3和r4各自独立地为氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基或c3-c7环烷氧基;
r3和r4可以位于h,i或j,但不能同时位于同一位置;
r5的每个实例独立地选自氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基,和c3-c7环烷氧基;
n是0,1,2,3或4;
x是nra,ch2,或-c(=o)-,其中ra的每个实例独立地选自氢和c1-c3烷基;
y1和y2各自独立地选自nra,ch2,和o;和
z是选自以下之一的基团的任何几何异构体:
和–c(=o)–。
在一个实施方式中,x不是–nh–和/或r3和r4不是异丙基。在进一步的实施方式中,如果z是–c(=o)–并且r3或r4是异丙基,则x不是–nh–,并且如果z是–c(=o)–并且x是–nh–,那么r3或r4不是异丙基。在另一个实施方式中,s1pr2拮抗剂可以是化合物1-7之一。如下所示的化合物1-7是抑制s1pr2的jte-013的类似物,并与jte-013相比具有改善的稳定性,所述jte-013描述在国际专利申请号pct/us2011/040637(wo2011/159864)中。
因此,在某些实施方式中,用于所公开的组合物、方法和试剂盒中的s1pr2拮抗剂选自:
在另一个实施方式中,s1pr2拮抗剂可以是通式ix的化合物,其中z不是–(c=o)–,特别地,s1pr2拮抗剂可以包含jte-013中的脲键(urealinkage)的生物电子等排体替代物或其类似物。
s1pr2拮抗剂也可以是由以下通式(x)表征的化合物:
其中,
r1是c1-c12烷基;
r2,r3,和r4各自独立地为氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基或c3-c7环烷氧基;
r3和r4可以位于h,i或j,但不能同时位于同一位置;
r5的每个实例独立地选自氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基和c3-c7环烷氧基;
n是0,1,2,3或4;
x和y各自独立地选自nra,o,和ch2,其中ra的每个实例独立地选自氢和c1-c3烷基;
和z是选自以下基团之一的任何几何异构体:
和-c(=o)-。
在一个实施方式中,z不是–c(=o)–。特别地,用于所公开的组合物、方法和试剂盒中的s1pr2拮抗剂可以选自以下所示的化合物8和9之一。
根据一些实施方式,s1pr2拮抗剂的治疗有效量的化学式选自下列化合物:2-[3-[(4-氨基-2-甲基嘧啶-5-基)甲基]-4-甲基-1,3-噻唑-3-鎓-5-基]乙基磷酸氢盐(pubchemidno.3382778),[(2s,3r)-2-氨鎓基-3-羟基十八烷基]磷酸氢盐(pubchemno.44317142)(也作为520和644260);(5ar,6r,9s,9as)-2-羧基-6-羟基-6-甲基-3-戊基-9-丙-1-烯-2-基-7,8,9,9a-四氢-5ah-二苯并呋喃(dibenzofuran)-1-醇盐(olate)(pubchemidno.54736865),2-(1-氨基-2-羟丙基)-n-癸基-1,3-噁唑-4-甲酰胺(pubchemidno.3866342),[(2s,3r)-2-氨鎓基-3-羟基十七烷基]磷酸氢盐(pubchemidno.46891770(也作为3247041),[(1s,2s,3s,4r,5r)-3-羟基-4-(4-甲基哌嗪-4-鎓-1-基)-6,8-二氧杂双环[3.2.1]辛-2-基]-(喹啉-3-基甲基)氨鎓(pubchemidno.51624406),5-[(2e)-2-(3-羧基-4-氧代环己-2,5-二烯-1-亚基(ylidene))肼基]-2-磺氧基苯甲酸(pubchemidno.9578291);5,7-二羟基-3-[3-羟基-4-甲氧基-5-(3-甲基丁-2-烯基)苯基]-2,3-二氢色满-4-酮(pubchemidno.9864156),5-羟基-2-(1-羟基-11-苯基十一亚基(phenylundecylidene))环己烷-1,3-二酮(pubchemidno.365015);3-[(3s)-3-氨鎓基-3-(2-羟基萘-1-基)丙酰基]-1-甲基-4-氧代喹啉(oxoquinolin)-2-醇盐(olate)(pubchemidno.28094480),2-[(e)-2-蒽-9-基-1-氰基乙烯基]-6-甲基喹唑啉-4-醇盐(pubchemidno.40592676),[(e,2s,3r)-2-氨鎓基-3-羟基十八碳-4-烯基]磷酸氢盐(pubchemidno.10883396),2-氯-3,6-二羟基-5-十一烷基环己-2,5-二烯-1,4-二酮(pubchemidno.342302),(2s)-2-氨基-2-(9h-芴-9-基甲氧基羰基)-3-羟基-4-甲基戊酸(pubchemidno.56923845),(5ar,6s,9s,9as)-2-羧基-6-羟基-6-甲基-3-戊基-9-丙-1-烯-2-基-7,8,9,9a-四氢-5ah-二苯并呋喃-1-醇盐(pubchemid.no.54734912);[(1s,2s,3s,4r,5r)-3-羟基-4-(4-甲基苯基)磺酰氧基-6,8-二氧杂双环[3.2.1]辛-2-基]-丙-2-基氨鎓](pubchemid.no.18390590),2-氨基-2-(9h-芴-9-基甲氧基羰基)-3-羟基-3-甲基丁酸(pubchemidno.56923928),[(1s,2s,3r,4r,5r)-3-羟基-4-(2-甲氧基乙基氨基)-6,8-二氧杂双环[3.2.1]辛-2-基]-[(4-苯基苯基)甲基]氨鎓(pubchemidno.51508548);(2-羟基苯基)甲基-[(1s,2s,3s,4r,5r)-3-羟基-4-苯基硫烷基-6,8-二氧杂双环[3.2.1]辛-2-基]氨鎓(pubchemid.no.28960354),[(1s,2s,3s,4r,5r)-3-羟基-4-(4-甲基哌嗪-4-鎓-1-基)-6,8-二氧杂双环[3.2.1]辛-2-基]-[(2-羟基苯基)甲基]氨鎓(pubchemid.no.51624683),(13-甲基-17-氧代-9,11,12,14,15,16-六氢-6h-环戊二烯并[a]菲-3-基)硫酸氢酯(pubchemidno.27993)。
适合用于本文公开的组合物、方法和试剂盒的化合物是拮抗s1pr2的化合物。s1pr2拮抗剂的非限制性实例包括本领域已知和描述的那些(参见,例如,国际专利申请号pct/us2013/033289(wo2013/148460)和pct/us2014/011033(wo2014/158302);us8,703,797;wo2011/159864;wo2008/154470;和wo2001/098301),以及与s1pr2结合口袋相互作用的本文鉴定的那些化合物(参见,例如,图8,9和11)。在某些实施方式中,也考虑了将拮抗s1pr2的这些化合物的类似物用于所公开的组合物、方法和试剂盒中。可以使用诸如本文所述那些和本领域已知的方法容易地测试s1pr2的拮抗作用。
根据一些实施方式,s1pr2拮抗剂可以是1-(2,6-二氯-4-吡啶基)-3-[(4-异丙基-1,3-二甲基-吡唑并[3,4-b]吡啶-6-基)氨基]脲,具有以下化学结构:
根据一些实施方式,s1pr2拮抗剂可以是由以下通式(i)表征的化合物:
其中:
-r1为c1-c12烷基,和r2,r3和r4各自独立为氢,卤素,c1-c6烷基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基或c3-c7环烷氧基,并且r3和r4任选位于h,i或j处,但不同时位于同一位置,且
-r5是卤素,c1-c6烷基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基或c3-c7环烷氧基,且n是0,1,2,3或4。
根据一些实施方式,s1pr2拮抗剂可以是以通式ii的通式为特征的化合物
其中
-x是nrarb,srb,f,ci,br或i,和
-r1是h或rb
-r2是h,f,ci,br,i,或rb
-ra是h或rb,且rb是具有1至12个碳原子的支链或直链烷基,其中一个或多个,优选1至7个氢原子可以被f,ci,br,i,ora,coor3,cn,n(ra)2替代,并且其中一个或多个,优选1-7个不相邻的ch2-基团可以被o,nra,s或s02,和/或被-ch=ch-基团替代,或者是具有3至7个环碳原子的环烷基或环烷基亚烷基,和
-w是c=0,c=s,s02或so,和
-q是nr3,-o-或-s-,和
r是氢,rb,ar或het,和ar是具有6-14个碳原子的单环或双环饱和,不饱和或芳族碳环,其可以是未被取代的、被下列基团单-,二-或三取代的:f,ci,br,i,rb,or3,-[c(r3)2]n-or3,n(r3)2,-[c(r3)2]n-n(r3)2,n02,cn,coor3,cf3,ocf3,con(r3),nr3coa,nr3con(r3)2,-[c(r3)2]n-het,-[c(r3)2]n-ar,-[c(r3)2]n-环烷基,-[c(r3)2]n-con(r3)2,-[(r3)2]n-coor3,-[c(r3)2]n-nr3-[c(r3)2]n-c02r3;-[c(r3)2]n-nr3-[c(r3)2]n-or3,-s02-[c(r3)2]n-c02r3,-s02-n(r3)2]n-[c02r3,-[c(r3)2]n-s02-[c(r3)]n-c02r3,-s02[c(r3)2]n-or3,-s02n(r3)2-[c(r3)2]n-or3,-[c(r3)2]n-s02-[c(r3)2]n-or3,nr3con(r3)2,nr3s02rb,cor3,s02n(r3)2,s02n(r3)rb,sorb,sonr3rb,s02rb,和/或-0[c(r3)2]n-coor3,并且het是具有1至4个n,o和/或s的单环或双环的饱和、不饱和或芳香族杂环,其可以是未被取代的、被以下基团单-,二-或三取代的:f,ci,br,i,rb,or3,-[c(r3)2]n-or3,n(r3)2,-[c(r3)2]n-n(r3)2,n02,cn,coor3,cf3,ocf3,con(r3),nr3coa,nr3con(r3)2,-[c(r3)2]n-het,-[c(r3)2]n-ar,-[c(r3)2]n-环烷基,-[c(r3)2]n-con(r3)2,-[(r3)2]n-coor3,-[c(r3)2]n-nr3-[c(r3)2]n-c02r3;-[c(r3)2]n-nr3-[c(r3)2]n-or3,-s02-[c(r3)2]n-c02r3,-s02-n(r3)2]n-[c02r3,-[c(r3)2]n-s02-[c(r3)]n-c02r3,-s02[c(r3)2]n-or3,-s02n(r3)2-[c(r3)2]n-or3,-[c(r3)2]n-s02-[c(r3)2]n-or3,nr3con(r3)2,nr3s02rb,cor3,s02n(r3)2,s02n(r3)rb,sorb,sonr3rb,s02rb,和/或-0[c(r3)2]n-coor3,和
-r1是h或rb,和
-r2是h,f,ci,br,i,或rb,和
-r3是h或rb,和
-n是0,1,2,3,4,5,6,7或8;
在某些实施方式中,所述化合物可以是选自以下的化合物:
2-[1-[2-(5-氯-2,4-二甲氧基-苯胺基)-2-氧代-乙基]-2,4-二氧代-喹唑啉-3-基]乙酸;-n-(5-氯-2,4-二甲氧基-苯基)-2-[2,4-二氧代-3-[2-氧代-2-[2-(3-吡啶基)乙基氨基]乙基]喹唑啉-1-基];2-[4-[1-[2-(5-氯-2,4-二甲氧基-苯胺基)-2-氧代-乙基]-2,4-二氧代-喹唑啉-3-基]苯基]-n-苯乙基-乙酰胺;4-[6-氯-1-[2-(3-氯-4-乙氧基-苯基)-2-氧代-乙基]-2,4-二氧代-喹唑啉-3-基]-n-环戊基-丁酰胺;n-(5-氯-2,4-二甲氧基-苯基)-2-[2,4-二氧代-3-[2-(苯乙基氨基)乙基]喹唑啉-1-基]乙酰胺;-2-[1-[2-(5-氯-2,4-二甲氧基-苯胺基)-2-氧代-乙基]-2,4-二氧代-喹唑啉-3-基]乙酸叔丁酯;-n-[2-[1-[2-(5-氯-2,4-二甲氧基-苯胺基)-2-氧代-乙基]-2,4-二氧代-喹唑啉-3-基]乙基]氨基甲酸叔丁酯;-2-[1-[2-(5-氯-2,4-二甲氧基-苯胺基)-2-氧代-乙基]-2,4-二氧代-吡啶并[3,2-d]嘧啶-3-基]乙酸;-2-[1-[2-(5-氯-2,4-二甲氧基-苯胺基)-2-氧代-乙基]-2-氧代-4h-喹唑啉-3-基乙酸;n-(5-氯-2,4-二甲氧基-苯基)-2-[3-(3-甲氧基苯甲酰基)-7-甲基-4-4a,8a-二氢-1,8-二氮杂萘-1-基]乙酰胺;2-[1-[2-[(2,6-二氯-4-吡啶基)氨基]-2-氧代-乙基]-5-甲基-2,4-二氧代-喹唑啉-3-基]乙酸;4-甲基-8-(2,4,6-三甲基苯胺基)-2h-酞嗪-1-酮;4-甲基-8-(2,4,6-三甲基苯胺基)-2h-异喹啉-1-酮;8-(2,6-二甲基苯胺基)-2h-异喹啉-1-酮;8-(4-氟-2,6-二甲基-苯胺基)-4-甲基-2h-酞嗪-1-酮;-4-乙基-8-(2,4,6-三甲基苯胺基)-2h-酞嗪-1-酮;4-异丙基-8-(2,4,6-三甲基苯胺基)-2h-酞嗪-1-酮;-4-(2-羟乙基)-8-(2,4,6-三甲基苯胺基)-2h-酞嗪-1-酮;-8-(2,6-二乙基-4-氟-苯胺)-4-甲基-2h-酞嗪-1-酮;8-(4-氯-2,6-二甲基-苯胺基)-4-甲基-2h-酞嗪-1-酮;-4-乙基-8-(4-氟-2,6-二甲基-苯胺基)-2h-酞嗪-1-酮;-5-(2-丙基吡唑基-3-基)2-2(2,4,6-三甲基苯胺基)苯甲酰胺;-5-甲氧基-2-(2,4,6-三甲基苯胺基)苯甲酰胺;5-氯-2-(2,4,6-三甲基苯胺基)苯甲酰胺。
在某些实施方式中,所述化合物可以是选自以下的化合物:
2-氨基-2-[2-(4-辛基苯基)乙基]丙烷-1,3-二醇;
5-[[3-氯-4-(2,3-二羟基丙氧基)苯基]甲基]-3-(邻甲苯基)-2-(丙氨基)噻唑烷-4-酮;
2-氨基-2-[2-[4-(3-苄氧基苯基)硫烷基]-2-氯-苯基]乙基]丙烷-1,3-二醇;
1-[5-[(3r)-3-氨基-4-羟基-3-甲基-丁基]-1-甲基-吡咯-2-基]-4-(对甲苯基)丁烷-1-酮;
3-氨基-4-(3-辛基苯胺基)-4-氧代-丁基]膦酸;和
5-[4-苯基-5-(三氟甲基)-2-噻吩基]-3-[3-(三氟甲基)噁二唑。
在另一个实施方式中,s1pr2拮抗剂可以是具有通式(iii)的化合物:
其中:
ar1是任选取代的杂环或芳族杂环;
ar2是任选取代的杂环或芳族杂环;
w是nra—,o,或—ch2—,其中ra是氢或c1-c3烷基;
z是—c(═o)—,—c(═s)—,o,—ch2—,═n—,或═ch—;
y是—nra—,—c(═o)—,—n═,—ch═,═n—,或═ch—;和
x是—nra—,—n═,—ch═,或—ch2—。
在另一个实施方式中,s1pr2拮抗剂可以是具有通式iv的化合物:
其中
ar1是芳香杂环;
w是nra—,o,或—ch2—,其中ra是氢或c1-c3烷基;
z是—c(═o)—,—c(═s)—,o,—ch2—,═n—,或═ch—;
y是—nra—,—c(═o)—,—n═,—ch═,═n—,或═ch—;和
x是—nra—,—n═,—ch═,或—ch2—;
r1是c1-c12烷基;
r2,r3,和r4各自独立地为氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基,或c3-c7环烷氧基;
r3和r4可以位于h,i或j,但不能同时位于同一位置;
x2是n或—crb—其中rb为氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基,或c3-c7环烷氧基。
在另一个实施方式中,s1pr2拮抗剂可以是具有通式v的化合物:
其中
r1是c1-c12烷基;
r2,r3,和r4各自独立地为氢,卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基,或c3-c7环烷氧基;
r3和r4可以位于h,i或j,但不能同时位于同一位置;和
r5的每个实例是卤素,c1-c6烷基,c1-c4烷氧基,c1-c6全卤代烷基,c1-c4全卤代烷氧基,氨基,单-或二-c1-c4烷基氨基,c3-c7环烷基,或c3-c7环烷氧基;
和
n是0,1,2,3,或4。
在某些实施方式中,在通式(iii)的化合物中:
r1是c1-c3烷基;
r2是c1-c3烷基;
r3在位置h,并且是c1-c6烷基;
r4是氢;
r5是卤素,和
n是2。
通式(vi),(vii)或(viii)中的另外的jte-013类似物,如下所示,具有s1pr2拮抗剂活性,描述于在美国专利号8,703,797中,其内容并入本文作为参考。
在另一个实施方式中,s1pr2拮抗剂可以是具有通式vi的化合物:
其中:
a是直接键或(cr)和b,c和d是独立地选自基团(cr)和n,其中r是h或烷基,然而条件是,并不是全部b,c和d是n,当a是直接键时,d是(cr);
r3选自烷基基团;
x选自o,nr4和cr4r5,其中r4和r5独立地选自h和烷基;
y选自o或s;和
z是取代的芳基环。
在另一个实施方式中,s1pr2拮抗剂可以是具有通式vii的化合物:
其中:
r1和r2独立地选自h和烷基,甲氧基,羟基,卤素,腈和三氟甲基;
r3独立地选自烷基,甲氧基,羟基,卤素,腈和三氟甲基;
d是cr或n;
r是h或烷基;
x是o,nr4,cr4r5,其中r4和r5独立地选自h和烷基,例如,低级烷基并且可以具有1至10个碳,和可以是具有3至10个碳的环状或支链烷基,甲氧基,羟基,f,br,i,腈和三氟甲基;
y是o或s,
z是取代的芳基环,具有以下结构:
其中r6和r7独立地选自烷基和可以包括1至10个碳,和可以是具有3至10个碳的环状或支链烷基,甲氧基,羟基,卤素,腈和三氟甲基;和
e是n或cr;
或,其中:
r1,r2和r3独立地为h,卤素,甲基或异丙基;
x是nr4;
r4是h;
y是o;
r6和r7独立地为h或氯;
e是n或cr;和
r是h。
在另一个实施方式中,s1pr2拮抗剂可以是选自以下的小分子:
n-(3,5-二氯苯基)-2-(4-甲基-1,8-二氮杂萘-2-基)肼甲酰胺;n-(3,5-二氯苯基)-2-(4-异丙基-1,8-二氮杂萘-2-基)肼甲酰胺;n-(3,5-二氯苯基)-2-(4-异丙基-5,8-二甲基喹啉-2-基)肼甲酰胺;n-(3,5-二氯苯基)-2-(4-异丙基喹啉-2-基)肼甲酰胺;n-(2,6-二氯吡啶-4-基)-2-(4,8-二甲基喹啉-2-基)肼甲酰胺;n-(3,5-二氯苯基)-2-(4,8-二甲基喹啉-2-基)肼甲酰胺;n-(2,6-二氯吡啶-4-基)-2-(4-甲基喹啉-2-基)肼甲酰胺;和n-(3,5-二氯苯基)-2-(4,5,8-三甲基喹啉-2-基)肼甲酰胺。
在另一个实施方式中,s1pr2拮抗剂可以是具有通式viii的化合物:
其中:
r1r2独立地选自h和烷基,甲氧基,羟基,卤素,腈和三氟甲基;
r3独立地选自烷基,甲氧基,羟基,卤素,腈和三氟甲基;
x是o,nr4,cr4r5,其中r4和r5独立地选自h和烷基,例如低级烷基且可以具有1至10个碳,和可以是具有3至10个碳的环状或支链烷基,甲氧基,羟基,f,br,i,腈和三氟甲基;
y是o或s;
r是h,甲氧基或烷基;
z是取代的芳基环,具有以下结构:
其中r6和r7独立地选自烷基并且可以包括1至10个碳,和可以是具有3至10个碳的环状或支链烷基,甲氧基,乙氧基,丙氧基,丁氧基,羟基,卤素,腈,和三氟甲基;e是n或cr;
或其中
r1,r2和r3独立地为甲基或异丙基;
x是nr4或cr4r5;
r4是h;
r5是h;
y是o;
r6和r7独立地选自烷基并且可以包括1至5个碳原子,甲氧基,乙氧基,丙氧基,丁氧基,氯和三氟甲基;
e是n或cr;和
r是h或甲氧基。
在另一个实施方式中,s1pr2拮抗剂可以是选自以下的小分子:
n-(3,5-二氯苯基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;1-(2,6-二氯吡啶-4-基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;n-(2-丁基-6-氯吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;n-(2-氯-6-乙氧基吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;1-(3,5-二氯苯基)-3-((1,3,7-三甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;n-(2,6-二氯吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;n-(3,5-双(三氟甲基)苯基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;n-(3-氯-5-甲氧基吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;1-(2,6-二氯苯基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;1-(2-氯-6-甲氧基吡啶-4-基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;n-(2-氯-6-丙基吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;1-(2-氯-6-丙基吡啶-4-基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;1-(2-氯-6-乙氧基吡啶-4-基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;1-(2-氯-6-丙氧基吡啶-4-基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;n-(2-氯-6-丙氧基吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;n-(2-丁氧基-6-氯吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;1-(2-丁氧基-6-氯吡啶-4-基)-3-((7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)甲基)脲;n-(2-乙氧基吡啶-4-基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺;和n-(5-氯-2,4-二甲氧基苯基)-2-(7-异丙基-1,3-二甲基-1h-吡唑并[4,3-b]吡啶-5-基)肼甲酰胺。
施用
通常将s1pr2拮抗剂化合物配制用于治疗用途。在某些实施方式中,本发明涉及药物组合物,其包含s1pr2拮抗剂化合物和药学上可接受的载体、稀释剂或赋形剂。药物组合物可以通过已知的方法使用众所周知且容易得到的成分制备(参见例如remington:thescienceandpracticeofpharmacy(20thed.),ed.a.r.gennaro,lippincottwilliams&wilkins,(2000)andencyclopediaofpharmaceuticaltechnology,eds.j.swarbrickandj.c.boylan,marceldekker,newyork(1988-1999))。
药物组合物可以通过任何合适的途径向受试者施用,例如全身通过静脉内注射、直接通过眼内注射、通过滴眼剂、口服等。组合物可以直接施用至靶位,通过例如手术递送至内部或外部靶位,或通过导管至血管可到达的部位。
例如,在治疗与sipr2有关的眼睛病症如fevr的方法中,可以通过眼内注射,滴注,口服或静脉内递送本文所述的组合物。所述组合物可以单次推注、多次注射或通过连续输注(例如静脉内、或通过腹膜透析经鞘内、泵输注)施用。对于胃肠外施用,组合物优选配制成无菌无热原形式。如上所述,本文所述的组合物可以是适于无菌注射的形式。为了制备这样的组合物,将合适的活性治疗剂溶解或悬浮于胃肠外可接受的液体媒介物中。在可接受的媒介物和溶剂中可以使用水,和葡萄糖溶液,1,3-丁二醇,林格氏液和等渗氯化钠溶液,通过加入适量的盐酸,氢氧化钠或合适的缓冲剂,将水调节至合适的ph值。水性制剂还可以含有一种或多种防腐剂(例如对羟基苯甲酸甲酯、对羟基苯甲酸乙酯或对羟基苯甲酸正丙酯)。在其中一种化合物仅少量或微溶于水的情况下,可以加入溶解促进剂或增溶剂,或者溶剂可以包含10-60%w/w的丙二醇等。本文所述的组合物可根据常规药学实践以任何合适的制剂施用于哺乳动物(例如啮齿动物,人类,非人灵长动物,犬科动物,猫科动物,绵羊,牛)(参见例如remington:thescienceandpracticeofpharmacy(20thed.),ed.a.r.gennaro,lippincottwilliams&wilkins,(2000)andencyclopediaofpharmaceuticaltechnology,eds.j.swarbrickandj.c.boylan,marceldekker,newyork(1988-1999),该领域的标准文本和usp/nf)。示例性的药学上可接受的载体和稀释剂以及药物制剂的描述可见于remington:同上。其他物质可以加入到组合物中来稳定和/或保存组合物。
本文所述的治疗方法通常包括将治疗有效量的本文所述组合物施用给有其需要的受试者(例如动物,人),包括哺乳动物,尤其是人。这样的治疗将适当地施用给受试者,特别是人,其患有、具有、易患疾病、障碍或其症状或处于疾病、障碍或其症状的风险中。可以通过任何客观或主观的测定,通过受试者或医疗保健提供者的诊断测试或意见来确定“处于风险中”的那些受试者。本文的方法和组合物用于治疗与贫血有关的任何其它病症或疾病。
有效剂量
本文所述组合物优选以有效量,即,能够在治疗的哺乳动物中产生期望结果的量(例如通过施用s1pr2拮抗剂治疗fevr),施用于哺乳动物(例如人)。这种治疗有效量可以根据标准方法来确定。
所述技术方法中使用的组合物的毒性和疗效可以通过标准的药物程序来确定。如在医学和兽医领域众所周知的,任何一个受试者的剂量取决于许多因素,包括受试者的尺寸、体表面积、年龄、要施用的特定组合物、施用的时间和途径、一般健康状况和同时服用的其他药物。如本文所述的组合物的递送剂量可以基于临床前有效性和安全性来确定。
实施例
以下具体实施例进一步说明了本技术。这些实施例仅用于说明,不应被解释为以任何方式限制本技术的范围。
通过s1pr2活性的基因抑制在fevr模型中的视网膜脉管系统的正常化
产生s1pr2-/-tspan12-/-双敲除小鼠,且观察到小鼠的视网膜脉管系统图式形成显著改善(图5)。(tspan12regulatesretinalvasculardevelopmentbypromotingnorrin-butnotwnt-inducedfzd4/beta-cateninsignaling.jungehj,yangs,burtonjb,paesk,shux,frenchdm,costam,riceds,yew.cell139,299-311(2009).)
还产生了fzd4-/-s1pr2-/-小鼠,并且这些小鼠也显示出视网膜脉管系统图式形成的改善(图6)。(vasculardevelopmentintheretinaandinnerear:controlbynorrinandfrizzled-4,ahigh-affinityligand-receptorpair..xuq,wangy,dabdouba,smallwoodpm,williamsj,woodsc,kelleymw,jiangl,tasmanw,zhangk,nathansj.cell116,883-95(2004))。因此,抑制s1pr2是对于fevr的显著治疗方法。
通过施用s1pr2拮抗剂在fevr模型中的视网膜脉管系统的正常化
用s1pr2拮抗剂jte-013对fzd4-/-小鼠进行产后治疗能够改善fevr视网膜血管化缺陷(图7)。fzd4-/-小鼠从p7到p25每隔一天腹膜内注射0.5mg/kg的jte-013。使用标准方案,将视网膜平放在p25处,并且在用异凝集素b4alexafluor594染色后,通过共焦显微镜可视化脉管系统。(vasculardevelopmentintheretinaandinnerear:controlbynorrinandfrizzled-4,ahigh-affinityligand-receptorpair..xuq,wangy,dabdouba,smallwoodpm,williamsj,woodsc,kelleymw,jiangl,tasmanw,zhangk,nathansj.cell116,883-95(2004).)。
计算机辅助s1pr2拮抗剂的药物设计
通常,g蛋白偶联受体被认为是高度可药用的,同时靶向s1pr1-3和-5的广谱特异性s1pr激动剂(芬戈莫德,商品名gilenya)在市场上用于治疗多发性硬化症。过去曾使用计算机辅助药物设计成功设计并合成小分子脂质酶抑制剂,其现在处于后续阶段1/2a临床试验的后期临床前评估。
为了通过计算手段鉴定s1pr2拮抗剂,使用分子操作环境(moe)程序对s1pr1结构进行建模。在整个过程中,charmm27力场得以实施,并规定了气相环境。从uniprot档案中获得s1pr2的氨基酸序列。将s1pr2和s1pr1的氨基酸序列进行比对,从比对产生同源性模型。产生的模型质子化温度为310k,ph值为7.0,盐浓度为0.1mol/l。moe中的站点查找工具被用来识别受体的结合口袋。识别出位于蛋白质的细胞外面上的由34个氨基酸组成的口袋(图8)。
如图9所示,s1pr2受体s1p的主要配体具有三个不同的化学区域。这些区域被用来在pubchem和先导物数据库中搜索含有区域1,2或3的类似分子。同样,将硫酸盐/酯(sulfate)基团也包括在搜索中,因为它们是磷酸盐/酯(phosphate)的生物电子等排体。根据以下标准鉴定化合物:它们必须具有小于390的分子量,区域2和3的xlogp值在-1和7之间,或xlogp值小于5,区域1总极性表面积为35-120。将来自每个区域所鉴定的化合物输入moe作为数据库。区域1共获得62,125个结果;区域2为2,971个,和区域3为13,442个。首先洗涤这些分子以除去可能已经包含在结构中的任何盐离子,并且使用charmm27力场在气相环境中进行能量优化。然后通过识别出的在s1pr2上的结合口袋对化合物进行虚拟筛选。
从虚拟筛选的结果中,选择每个区域最好的100种化合物,并采用更严格的对接方法:诱导拟合s1pr2和s1pr1。该对接使得衬于口袋的氨基酸侧链移动,以及配体停靠。对得到的数据库进行s分数优于s1p分数的化合物的检测,并预测了s1pr2与s1pr1的特异性。然后筛选可购买的经鉴定的化合物,并且发现36种化合物市售可得,并被选作用于测试的有活力的靶标。如图11a-d所示,来自pubchem的使用pubchem识别号的是以下那些:3382778;44317142(也作为520和644260);54736865;3866342;46891770(也作为3247041);51624406;9578291;9864156;365015;28094480;40592676;10883396;342302;56923845;54734912;18390590;56923928;51508548;28960354;51624683;27993。
它们的有效性可以容易地与jte-013相比较,所述jte-013是具有选择性s1pr2抑制特异性的s1pr拮抗剂。
fevr的斑马鱼模型中s1pr2拮抗活性的示范和排序
敲除方法允许重现人类疾病的斑马鱼模型的产生,在更费时和昂贵的哺乳动物研究之前,允许对药物筛选的快速中间体体内步骤。s1pr2药物靶标和fzd4通路在斑马鱼、小鼠和人之间高度保守。使用talen系统产生胚系fzd4-/-斑马鱼(图10)。
为了测定经鉴定的s1pr2拮抗剂以及已知的工具化合物jte-013,将三个fzd4-/-斑马鱼胚胎排列在96-孔板中。在受精后24小时(hpf),将化合物以0.01-30μm的终浓度转移到胚盘中。然后将胚胎与化合物在28.5℃温育12小时并筛选,用于总的总体发育效果。然后在不同的时间范围(2-12天),将胚胎用三卡因(ms-222)过量处理,并用4%多聚甲醛固定,并用显微镜确定其视网膜脉管系统。随后在tspan12-/-和fzd4-/-小鼠中测试那些使斑马鱼最佳恢复正常脉管系统的化合物以分离最有效的治疗化合物。
对于在小鼠中的工作,通过眼内注射将化合物递送至p17和p28之间的小鼠眼睛(0.01-30μm),其可以通过s1pr2拮抗剂有效治疗。该时间范围类似于fevr在人类中发生的阶段。然后如上所述确定对于tspan12-/-和fzd4-/-fevr小鼠模型和其他fevr遗传模型研究的视网膜表型和眼功能。
- 还没有人留言评论。精彩留言会获得点赞!