分子质量保证方法在测序中的应用与流程
本发明涉及质量保证方法。
背景技术:
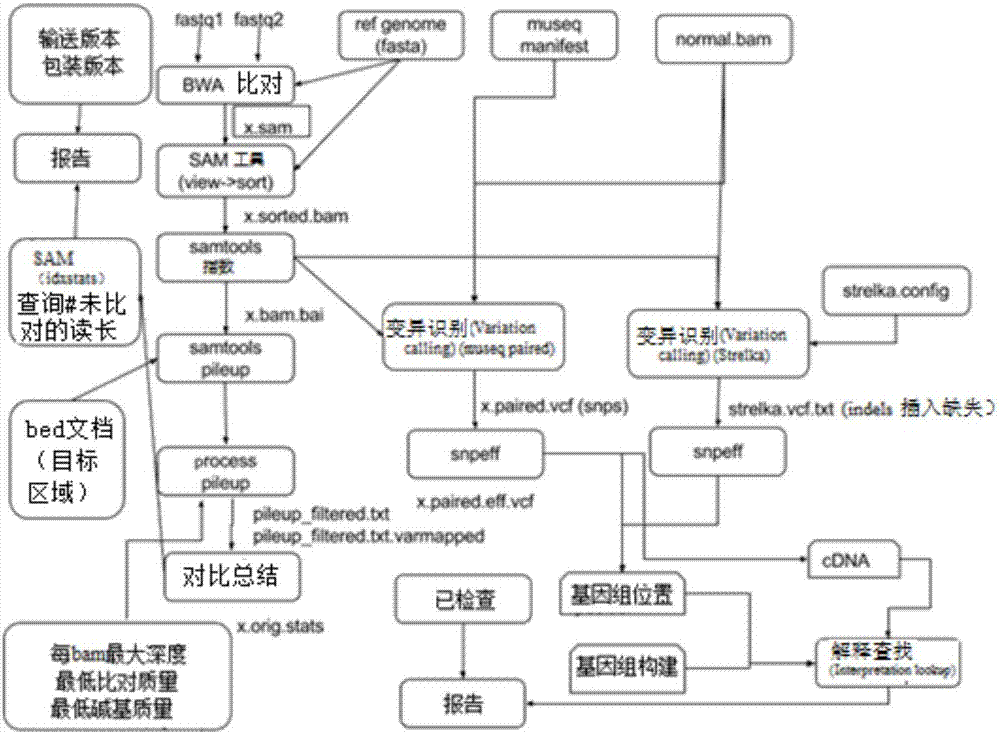
:数字单分子基因组测序通常被称为下一代测序技术(NGS),它通过合成方法进行测序,其与单分子DNA测序相近。NGS方法的一个特点是它们代表在序列中衍生的单个分子。在基于基因组的癌症测定中,NGS用于基因组分析。然而,NGS的几个方面将从建立等位基因识别(allelecalls)的置信度的质量保证过程中受益。这些方面包括对等位基因扩增中生物方面和技术方面偏差的检测、对测序中不良模板或用量不足的模板的检测、对外来的扩增子污染的检测、以及对输入DNA池过程中低发的(lowprevalence)真正的突变的检测。质量保证是临床试验的必要因素,也有助于形成健全的研究基础。已有几种方法用于对DNA分子进行检测,例如,采用DNA序列的随机连接,其中碱基序列代表一个字符(word)或代码(code)(称为条形码,或分子条形码),然后进行扩增。已知的DNA代码方法的局限性在于,它们一般不能对用于检测的有偏差的代码分子集带来的结果进行处理,也不能对依赖于序列的在连接效率上的损失带来的结果进行处理。此外,还需要一些方法来将分子检测与NGS序列的等位基因检测的概率方法结合起来(例如那些采用贝叶斯图形模型的方法,如SNVmix(1),并将其纳入基于特征的序列变异分类器中,如mutationseq(2)。)技术实现要素:一方面,本公开提供了一种确定核酸模板的复杂度的方法,包括以下步骤:i)提供一个核酸模板;ii)提供多个引物对,其包括第一引物和第二引物,其中所述第一引物包含与所述核酸模板的一部分互补的序列,所述第二引物包含与所述核酸模板的互补序列的一部分互补的序列;iii)将代码连接到所述第一引物的5’末端、所述第二引物的5’末端或两者之上,以形成代码-引物分子,或将代码连接到所述核酸模板上以形成代码-模板分子;iv)用所述配对的代码-引物分子和核酸模板,或用所述引物对和代码-模板分子进行确定循环数的扩增反应,以获得扩增反应产物;v)在每个循环结束时、在确定的循环数结束时、或在任一中间的循环数时,获得扩增反应产物的序列;vi)在每个循环结束时、在确定的循环数结束时、或在任一中间的循环数时,确定扩增反应产物中每个代码的丰度;vii)确定每个循环所观测的代码熵(codewordentropy);以及viii)将观测的代码熵与估算的代码熵进行比较,以确定核酸模板的复杂度。另一方面,本公开提供了一种识别真正的序列变体的方法,包括以下步骤:i)提供一个核酸模板;ii)提供多个引物对,其包括第一引物和第二引物,其中所述第一引物包含与所述核酸模板的一部分互补的序列,所述第二引物包含与所述核酸模板的互补序列的一部分互补的序列。iii)将代码连接到所述第一引物的5’末端、所述第二引物的5’末端或两者之上,以形成代码-引物分子,或将代码连接到所述核酸模板上,以形成代码-模板分子;iv)用所述配对的代码-引物分子和核酸模板,或用所述引物对和代码-模板分子进行确定循环数的扩增反应,以获得扩增反应产物;v)在每个循环结束时、在确定的循环数结束时、或在任一中间的循环数时,获得扩增反应产物的序列;vi)在每个循环结束时、在确定的循环数结束时、或在任一中间的循环数时,确定扩增反应产物中每个代码的丰度;vii)确定每个循环所观测到的代码熵;以及viii)根据步骤vi)和vii)的结果,应用监督分类方法来识别真正的序列变体。真正的序列变体可以是低发的序列变体。核酸模板可以是DNA模板。代码-引物分子或引物可进一步连接到接头序列(adaptersequence)上。连接到引物对的第一引物和第二引物上的代码可以是不同的,或连接到引物对的第一引物和第二引物上的代码可以是相同的。代码可以随机连接到核酸模板上。所观测到的代码熵可以由多样性指数,如Shannon熵、Simpson指数或任何其他多样性指数来计算。代码可存在于非均匀的池中。代码可存在于本文所述的均衡池中。本文所描述的方法可用于检测真正的序列变体,扩增过程污染,样本一致性错配或代码池不均衡。另一个方面,本公开提供了一种获得均衡的代码池的方法,包括:i)提供包括具有定义长度的多个代码的初始样本;ii)提供目标序列;iii)提供多个引物对,包括第一引物和第二引物,其中,第一引物包括与目标序列的一部分互补的序列,第二引物包括与所述目标序列的互补序列的一部分互补的序列,其中,每个代码连接到第一引物的5’末端、第二引物的5’末端或两者之上,以形成配对的代码-引物分子;iv)用所述配对的代码-引物分子和目标序列进行确定循环数的扩增反应,以获得扩增反应产物;v)在每个循环结束时、在确定的循环数结束时、或在任一中间的循环数时,获得扩增反应产物的序列;vi)在每个循环结束时,在设定的循环数结束时,或在任一中间的循环数时,确定扩增反应产物中每个代码的丰度;vii)获得代码性能的测量参数,包括以下步骤:a)将步骤(vi)的丰度与预期值进行比较;和/或b)确定每个在前扩增循环中丰度增加的速率;并利用步骤(vii)的测量参数,采用随机局部搜索方法在电脑上(insilico)执行搜索,以获得均衡的代码池。代码-引物分子可以进一步连接到接头序列(adaptersequence)上。代码长度可以是约4个单位至21个单位。初始样本大小可以是至少10个代码。初始样本可以是随机样本,也可以是经组合的和/或热力学约束的(thermodynamicconstraints)。初始样本可以包括代码序列的所有组合,或可以包括代码序列组合的子集。可以使用较大的代码池或不同长度的代码池来执行该方法。可以使用单个目标序列或使用两个或多个目标序列执行该方法。该方法可以执行一次,两次或两次以上。该方法可以包括对作为子序列和位置函数的代码性能的确定。引物可以包括SEQIDNOs:1-146中所示的一个或多个序列。在某些方面,本公开提供了一组引物对,包含第一引物和第二引物,其中所述第一引物包含SEQIDNOs:1-73中所示的任一序列,所述第二引物包含SEQIDNOs:74-146中所示的任一序列。在一些实施方式中,引物或引物对可与适于存储、运输、递送、或使用引物或引物对的试剂一起制备试剂盒,任选地所述试剂盒还包含使用说明书。本发明概要不一定描述了本发明的所有特征。附图说明结合以下对附图的描述,本发明的这些特征和其他特征将会变得更加清楚,其中:图1是表示患者样本工作流程的流程图;图2是表示序列分析工作流程的流程图;图3是一个矩阵,将代码性能表示为子序列的组成和位置的函数;图4是一种确定对代码性能影响较大的参数的算法;图5是用于NGS测序反应的DNA模板和引物的示意图;图6是在一个示例性测序反应的前4个PCR循环中观测到的扩增序列和代码的示意图。该图表显示了在前3个PCR循环中包含的所有代码。然而,只有扩增序列的代码显示在第4个PCR循环中。图7是在扩增过程中添加代码的机制的示意图;图8是i~U(1)的第4个PCR循环中代码熵的分布的箱形图;图9是表示i~U(1)和i~U(3)的第4个PCR循环中代码熵分布的对比箱形图,其中x轴上的标签对应于用于产生每个分布的参数(例如,u1_m1对应于i~U(1)并且m=1);图10是显示代码多重数变化的泊松分布(Poissondistribution)模型的图,其中实线曲线对应于随机生成的泊松分布i~P(λ=6),其中和虚线曲线具有相同的分布,其中值移位1(在这种情况下图11是i~U(1),i~P(λ),其中,λ=1,3,6,m=1..10时的第4个PCR循环中代码熵的分布的对比图;图12是i~U(1),i~P(λ),其中,λ=1,3,6,m=300,1000,2000,3000时的第4个PCR循环中代码熵的分布的对比图;图13是表示第4个PCR循环中每个代码wj出现的概率的图,其中当i~U(1)时当i~P(λ)时图14是代码多重数变化的负二项分布模型,其中实曲线对应于随机生成的负二项分布i~NB(r=6,p=0.5),其中虚线曲线具有相同的分布,其值移位1。在这种情况下图15是当i~U(1),i~NB(r,p)时m=3000时第4个PCR循环中的代码熵分布的对比图,其中x轴上的标签对应于用于生成每个分布的参数(例如,nbinomial1_p.1对应于移位分布i~NB(r,p=0.1)+1,此时μ=1。也就是);图16是图15所示的平均熵与负二项分布方差之间的关系图;图17A是当i~NB(r,p)和m=1时的代码熵的分布图;图17B是当i~NB(r,p)和m=5时的代码熵的分布图;图17C是当i~NB(r,p)和m=10时的代码熵的分布图;图18A显示了图17A所示的负二项多重数分布的方差与熵分布的平均值的相关性;图18B显示了图17B所示的负二项多重数分布的方差与熵分布的平均值的相关性;图18C显示了图17C所示的负二项多重数分布的方差与熵分布的平均值的相关性;图19是对于i~U(1)和具有离群值的i~U(1)的第4个PCR循环中的代码熵分布的对比图。异常值的数量在2至70之间,随机多重数在5至7之间变化。在每种情况下,模板分子的初始数目是m=5,池中unique代码的总数是14*m=70;图20是表示2个,3个和4个PCR循环以及不同数量的初始模板分子m的代码熵的分布图;图21A是表示扩增产物的熵位于具有相应初始模板分子浓度的预期的熵的分布的情况图。图21B表示当PCR过程的扩增产物的熵具有较低的值时,说明有伪差(artifact)存在的情况。图22是使用代码熵来评估扩增产物的质量的示意图;图23A是表示具有和不具有代码(m=5000)的扩增子性能的图;图23B是表示具有和不具有代码(m=10000)的扩增子性能的图;图24A是表示熵作为8-mer起始模板数的函数的图,其中熵是根据包含5号染色体136633338位置的给定等位基因的所有读长来计算的;图24B是表示熵作为10-mers起始模板数的函数图,其中熵是根据包含5号染色体136633338位置的给定等位基因的所有读长来计算的;图25是表示若干数目的起始模板的代码熵的分布图,其中熵是根据来自属于相同扩增子的读长的所有代码来计算的;图26是次要SNP等位基因的代码熵作为模板初始数的函数图;图27是伪突变的等位基因(artifactalleles)的代码熵作为模板初始数的函数图;以及图28是伪突变的和真正突变的变体的熵的图,其中标签中显示了训练集和测试集以及所有真正突变的%VAF。具体实施方式某种程度上,本公开部分地提供了用于确定均衡性能代码池的相关序列参数,并且从一开始利用所测量的参数来设计较大的均衡池的方法。核酸代码的分子检测池(如DNA或RNA)可用于对起始模板数、质量和检测/避免PCR/测序/DNA合成错误的进行评估。可采用熵的方法和相关信息论的方法,例如来确定模板数和错误控制,从而对随机引入的核酸代码的检测进行分析。一方面,本公开提供了用于设计和选择用于随机连接至目标序列或模板(例如核酸模板)的合适的代码池的方法。所谓“核酸模板”或“目标序列”是指DNA,RNA或DNA/RNA杂交分子或互补分子。核酸模板或目标序列可以分离自样本,包括但不限于临床样本,生物学研究样本或法医样本,或者可以是人工序列,例如合成序列或重组序列。在一些实施方式中,核酸模板或目标序列包括但不限于临床或生物学意义上的序列,如患者实体瘤或循环无细胞DNA样本的体细胞突变热点,或法医感兴趣的序列。在一些实施方式中,核酸模板或目标序列包括但不限于包含突变的序列(“真正的序列变体”)。真正的序列变体可以包括低发的真正的突变,例如变异等位基因频率(VAF)小于1%、2%、3%、4%、5%、6%、7%、8%、9%或10%的突变。在一些实施方式中,并不普遍的真正的突变的VAF可小于5%。所谓“互补”是指两种核酸,例如DNA或RNA,其含有足够数量的核苷酸,能够形成Watson-Crick碱基对,从而在这两种核酸之间形成一个双链区。因此,DNA或RNA的一条链中的腺嘌呤与互补DNA链中的胸腺嘧啶或互补RNA链中的尿嘧啶配对。可以理解的是,并非核酸分子中的每个核苷酸都需要与互补链中的核苷酸形成匹配的Watson-Crick碱基对以形成双链结构(duplex)。核酸模板或目标序列可以是任意长度的或由任意核苷酸组成的,如具有两个或多个共价键合的核苷酸的任意链,包括自然存在或非自然存在的核苷酸,或核苷酸类似物或衍生物。随机生成的代码池可以足以用于熵评估,但是随机生成的代码集可以包含在PCR测序反应中表现较差的核酸序列,从而减少用于对模板分子进行检测的信息内容,或对该信息内容造成偏差。因此,在一些实施方式中,测量扩增的起始模板之间的熵的差异,对于优化性能是有用的。一方面,本公开提供了一种获得均衡代码池的方法。所谓“代码”是指一种线性聚合物分子,其具有唯一确定的序列,例如不限于是DNA,RNA,DNA/RNA杂交链(DNA/RNAhybrid)或其他能够被扩增的大分子。虽然本文列举的方法是指DNA分子,但要理解的是,这些方法普遍适用于其他能够被扩增的分子。代码可以是长度“k”。长度k可以定义为任意长度,比如至少4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21个单位(例如核苷酸碱基或氨基酸残基)或更长,尽管渐增的较大长度可能导致成本增加和效率下降。在一些实施方式中,长度k可以是10。所谓代码的“均衡池”是指允许均衡的热力学设计以避免偏差扩大或代码并入的代码池,和/或足够独特以容忍代码一致性确定中的测序错误的代码池。合适的均衡代码池中的代码可以按照|W|≈m*(2c-2)进行排序(其中m是模板的初始数,c是PCR循环数),以便用于对本文所述的熵进行评估。一般来说,在不受任何特定理论约束的情况下,平衡的代码池提供均衡的性能,并且可能能够对类似的扩增的性能进行区分。在一些实施方式中,提供了具有定义长度k的多个代码的初始样本。代码的初始样本可以表示序列的所有组合或其子集,例如,超过10或超过100的不同的代码,但是应当理解的是,池的大小将限制可能的组合。在一些实施方式中,代码的初始样本可以与用于测试的池的大小相同。可以使用任何合适的技术来生成长度为k的代码序列组合,例如通过在代码序列中引入一系列Ns(即A、G、C、T或U)的方式来引入随机碱基,或通过所有长度为k的代码子序列的明确规范的组合,提供给寡核苷酸合成器,或是两种方法的结合。这些技术对于本领域技术人员来说是熟知的。在一些实施方式中,可以使用修饰碱基,例如包含硫基的碱基或其他修饰碱基。在不受任何特定理论约束的情况下,修饰碱基可以改变代码的热力学性质,或可以提供一种通过物理方法检索代码的方法,例如加入生物素部分,以用于生物素-链亲和素的捕获。与代码性能有关的序列特征参数在一些实施方式中,一个或多个以下组合的和/或热力学约束可应用于代码。在本文描述的方法中,W是定义为长度为k的核苷酸碱基的线性序列的代码w的集合。即在物理现实中,每个条形码DNA序列或代码可以包括编码该序列的多个相同的分子。因此,寡核苷酸物理池中的代码分子的多重集可以定义为M={w:i|w∈W和i=1,2,..,其中w是根元素(rootelements),而i=i(w)是w的重数(multiplicity)。也就是说,w的重数是在多重集M中观测到的w的实例数。根集(unique代码)的基数(cardinality)是|W|=p,而多重集M的基数为通过引入组合的和热力学约束,可以对高质量池M的设计进行建模。高质量的代码不会减少扩增的DNA模板序列的数量。可以在根元素w上施加一个或多个组合约束,其中H是代码对(wi,wj)的代码间距(Hammingdistance),其被定义为两个相同长度wi和wj的代码完美比对时的错配数。C1:代码错配(HD_w)。H(wi,wj)≥dw与wi,wj∈W。执行池中所有可能的代码对之间的大量错配。C2:代码基因组错配(HD_g)。人类基因组中发现的H(wi,wg)≥dg与wi∈W和k-merwg。为避免在PCR过程中所述代码与人类k-mers相互作用,在模型中引入了每个代码与所有人类k-mers之间的dg不匹配。C3:标记的引物基因组错配(HD_gp)。wip中所有的k-mer子序列ws,定义为结合有引物p,具有H(ws,wip)≥dp与wip∈W。该约束确保了带有引物序列(containerprimersequence)的代码边界不会在基因组中产生偶然的同源性(inadvertenthomology)。C4:标记的引物对错配(HD_pp)。代码标记的引物。这种约束确保了代码标记的引物之间不发生相互作用。C5.GC含量。每个wj∈W的GC含量c为45≤c≤60。代码的稳定性和一致性可以通过检测在同一个代码内特定的碱基G和C来建模。还可以施加下列一个或多个热力学约束来避免不期望有的相互作用。T1.发夹解链温度。对于每个代码及引物wip,所有可以潜在地与序列wip形成的发夹的最高解链温度必须低于temp_hairpin。发夹的形成在PCR过程中能阻止条形码标记的引物对DNA模板的退火。T2.自二聚体自由能。与引物wip连接的每个代码的二级结构的自由能ΔG(wip)必须大于阈值ΔGdimer。该约束能够禁止wip二级结构的形成,从而防止条形码标记的引物对DNA模板的退火。T3.异二聚体自由能。由两个条形码标记的引物wip(i)和wjp(j)相互作用形成的异二聚体的自由能ΔG(wip(i),wjp(j),必须大于所有wip(i)和wjp(j)的阈值ΔGheterodimer。该约束条件能够禁止在成对的条形码标记的引物之间形成能够避免DNA模板退火的二级结构。对于确定的代码长度,根集W的大小随约束数而减小。然而,所需的unique代码的数量随着PCR循环数和DNA目标模板的质量而增加。例如,在一个反应中,模板分子的绝对数量可以通过单倍体人类基因组的质量估测至约3.4pg(即3x10-12g)。一个典型的目标PCR测序反应将使用1ng至10ng的模板分子,即每个单倍体目标位点在~300至~3000个拷贝之间,或者是该数值的两倍,即每个二倍体在~600至~6000拷贝之间。然而,本文所描述的方法可以确定直至单个模板分子的熵。对于4个PCR循环,当考虑上文所描述的设计约束C1-C5和T1-T3时,一个目标位点的每一端需要300*14至3000*14代码。然而,将池设计为每个目标位点和每一端具有不同的代码集。也就是,其中LiR和LiF是目标位点Li的反向末端和正向末端。因此,在含有x目标位点的实验中,所需的不同的代码数为300*14*2*c=8400*c至3000*14*2*c=84000*c。因此,大量的多样的代码集是有用的。可以使用更长和更短长度的代码,具体取决于C1-5和T1-3中所示的所需约束条件。然而,施加于代码上的约束应当是所需的最小值,以避免不期望有的交互作用,并且同时确保unique代码的数量足够大,以在4个或更多的PCR循环中获得高的代码熵。对代码子样本上的代码性能参数的测定在一些实施方式中,可以使用穷举法对所有固定长度的代码进行物理测试,并选择在各种应用中能产生最佳PCR扩增的代码,以便确定对扩增效率有较高影响的代码特性(例如,C1-5和/或T1-3中的一个或多个)。在可替代的实施方式中,对于例如长度为4至21个碱基的代码,如4、5、6、7、8、9、10、11、12、13、14、15、16、17,18、19、20或21,可以使用减少特征选择空间的方法。采用用于特征选择的Lasso(最小绝对选择和收缩算子(LeastAbsoluteSelectionandShrinkageOperator))方法来确定产生相似代码性能的特征。该方法通过对回归中所得到的权重的L1范数的惩罚来拟合线性模型。该系数估算为其中yj是响应变量或代码性能,Xj是解释变量(explanatoryvariables)或特征,λ是分配给每个代码属性βj的权重。调谐参数λ控制惩罚的强度。即,是λ=0以及当时的线性回归估测值。交叉验证可以用来选择λ的最佳值。应当理解的是,任何其他特征选择方法,或诸如AdaBoost的分类方法,都可以用来确定对扩增效率有较大影响的代码特性。在一个示例中,生成了代表长度定义为k的序列或其子集的所有可能组合的代码的初始样本。在一些实施方式中,代码的初始样本包括至少10个不同的代码。在可替代的实施方式中,代码的初始样本包括100多个不同的代码。在一些实施方式中,如果测量长度为k的代码的完整集,这可以被视为长度为k+1,k+2等代码的子集。在一些实施方式中,其中k是10,可以生成所有可能的代码序列组合。一般来说,代码的初始样本应该与长度k成比例,以获得一组具有代表性的代码。在代码的初始样本中,每个不同的代码都可以连接到单个目标序列引物或引物对的5'末端,从而形成代码-引物分子。所谓“引物对”是指两个优化设计的寡核苷酸序列(“第一引物”和“第二引物”),如正向引物和反向引物,可用于引导聚合酶链式反应,其中第一引物和第二引物与目标序列的任意一条链上的互补序列进行退火。引物对中的一条引物可以具有任意适宜的长度,例如至少8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25个核苷酸碱基或更长,尽管越来越长的长度可能导致成本增加、合成错误或效率下降。在一些实施方式中,引物对中的一条引物可以是15个核苷酸碱基。在一些实施方式中,相同的代码可以连接到引物对的第一引物和第二引物上。在可替代的实施方式中,不同的代码可以连接到引物对的第一引物和第二引物上。在一些实施方式中,代码只能连接到引物对的一条引物上。在可替代的实施方式中,代码可以连接到引物对的两条引物上。可以使用任何合适的技术,例如寡核苷酸合成或连接(ligation)或其他合适的方法,将代码连接到引物上。例如,长度为k的所有可能的代码的初始样本在单个目标序列引物的5’端合成(如本文所描述的CG001v2panel序列中披露的位点引物对,见表15)。在一些实施方式中,如图5和本文描述的CG001v2分析中所述的,用于PCR产物库构建的接头序列可以作为5’末端到代码的合成的一部分。接头序列可以是核酸序列,例如DNA序列,专门设计用于在NGS平台上进行的测序化学反应,其中测序库分子被连接到玻璃液流细胞表面或磁珠上,并从分子的任一端连续进行核苷酸碱基鉴定。接头序列在本领域中是已知的,许多这样的序列是商业可得的。一个目标序列,包括与每个目标序列引物对的序列互补的序列,可以使用代码-引物分子对通过任何适当的扩增反应来扩增,例如聚合酶链式反应(PCR)或任意使用任何能扩增核酸链的聚合酶的合适的线性扩增技术,依次应用,例如不限于T4聚合酶、phi29聚合酶或逆转录酶(在RNA情况下),以提供包括代码序列在内的扩增反应产物。扩增反应产物的序列可以使用任何合适的技术来获得,包括但不限于,在玻璃流动细胞上进行合成测序、在磁珠上进行焦磷酸测序的下一代DNA测序化学,或质子半导体技术,再结合上核苷酸碱基读数作为光信号或离子pH的变化。其他正在使用的技术包括利用纳米微孔(nanowells)和纳米孔(nanopores)的真正单分子实时测序。在一些实施方式中,代码的扩增性能可以由如下方法确定。PCR目标反应可以使用,例如本文所述的CG001v2分析过程来进行。然而,可以预先确定扩增循环数(设定的循环数)来使反应停止,以确定代码丰度的增加速率。因此,可在4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35或更多的循环数,或在这些循环的子集(中间循环数)的任意组合中,获取代码-目标PCR反应的样本。在一些实施方式中,可以使用巢式PCR技术来执行额外的扩增循环。在一些实施方式中,界线c*可以由PCR反应的循环数来确定,尽管c*循环需要的预期代码数应该最多是代码池的大小。然后,在每个循环结束时,在设定的循环数结束时,或在中间循环数结束时,可在任何下一代测序(NGS)装置或任何能够提供核酸模板序列数字检测的设备上(例如在CG001v2分析中所述的或本领域技术人员熟知的任何PCR-NGS测序方法),对PCR反应编索引和测序。在每个循环结束时,扩增反应产物中代码的丰度可以通过,如DNA序列比对和代码实例检测(如CG001v2分析中概述的(图2))来确定。可以使用不同的策略提取代码。例如,通过比对一组引物与扩增子测序数据,并修剪引物和5‘端之间发生的k-mers。该实用程序可以支持在匹配引物序列时设置代码间距阈值。为了获得高质量的数据,配对的两个读数均需经过过滤器。此外,诸如引物二聚体之类的低质量读数可以使用额外的度量标准(例如,根据成对读数的成对序列对比的互补碱基的数目来计算的编辑距离)来过滤掉。扩增子中,来自编辑距离大于所有读数的模式编辑距离阈值的读数的代码,可以被过滤掉。这显示了长度为k(在每个PCR循环中表示为4至35)的代码的数量。然后,代码序列的性能可以通过(i)由该方法的一次或多次迭代得到的观测到的和预期的代码丰度之间的关系,和/或(ii)随PCR循环数增加的丰度增长率来计算。另一种方法可以是(iii)分析观测到的代码频率的分布。对于(i)来说,在假设熵服从正态分布的前提下,利用预期的熵分布的参数来计算观测到的熵的z-得分值,来得出在预期的熵分布的条件下观测到的熵的概率。可以使用其他统计方法将观测到的熵与预期的熵分布进行比较,这对于本领域技术人员来说是熟知的。(ii)中的代码扩增系数可以直接或通过线性建模进行计算,例如,给定字符Yw的丰度被建模为β0+β1*X的函数,其中X是PCR循环数,β1是扩增系数的估测值。β0的值与第一次观测到代码w的循环有关。PCR的序列扩增是指数型的,但代码扩增是线性的(图6和表1)。表1.每个PCR循环的代码频率因此,在perfectPCR反应中,Yw=β0+X作为代码预期每个PCR循环增加一个。对于(iii),所观测到的代码频率分布可用于识别扩增性能差的代码或优先扩增的代码。观测到的频率值应在[a,b]的范围内,由于代码扩增是线性的,而且在PCR反应的后期循环中会引入更多的代码,因此频率为i的代码数应当等于或高于频率为j的代码数,其中a≤i<j≤b。过度扩增的一个例子是,当频率没有在[k,b-1]范围内的代码时,其中a<k<b,但观测到有一个代码的频率b远高于其余观测到的频率。也就是k<<。在这种方法中,因为在一个测试的数以亿计的扩增读数中,只有一部分被测序,因此包含给定代码的整个读数样本中,只有一小部分可以被观测到。用于优化性能指标的迭代过程在上述示例中,对于确定长度的代码,确定了有利和不利的代码属性。在一些实施方式中,可以生成更短或更长的代码(例如,1、2、3、4或更多的连续代码长度),以提供额外的性能指标,并且可以使用不同长度的代码重复扩增步骤和分析步骤。图4显示了对在PCR扩增中具有较大影响的热力学的和序列参数的迭代过程。在一些实施方式中,扩增步骤和分析步骤可以在单个目标位点上执行。在可替代的实施方式中,可以在2个或更多位点上执行扩增步骤和分析步骤,通过连接到单个目标位点序列上的代码-引物分子的性能,来评估目标位点特定序列的独立性。在一些实施方式中,对于每个代码长度和/或目标位点来说,可以对整个过程(代码的生成、扩增和分析)执行一次。在可替代的实施方式中,对于每个代码长度和/或目标位点来说,可以对整个过程(代码的产生、扩增和分析)重复执行两次或两次以上。在一些实施方式中,可确定重复测量之间的方差,并且在方差低于期望值如1%、5%、10%、15%、20%、25%等时,停止重复测量。需要了解的是,技术人员会很容易地认识到测量值在某一特定值附近稳定的点,并在此点之后不再继续重复。作为序列组成函数的代码性能的测定在确定的目标位点测定确定长度的代码的性能后,与性能相关的参数可按如下所述进行确定。在一些实施方式中,代码性能可以被归类为子序列的组成和位置的函数。关于有利的和不利的子序列的组成和位置的信息可以用来设计更长的更可能表现出良好的PCR扩增的代码。在一些实施方式中,可按如下所述检测影响PCR扩增的代码的子序列。设Wk为长度为k的代码的集合。即,其中Wk的大小是|Wk|=4k。在PCR反应中,测定每个代码或代码子集wj∈Wk的指标yj。然后用行中的子序列组成和列中的子序列位置生成矩阵。矩阵的元素是具有特定子序列组成和位置的代码的中值指标。例如,在图3所示的矩阵Y中,第一行对应的是子序列为AA的代码,第一列对应于在代码的第一和第二位置中发现的均聚物。因此,y11是所有代码w=AAw3..wk(其中,wi∈{A,G,C,T}i=3…k)的中间扩增(medianamplification)。矩阵中的子序列具有固定长度,因此为每个可能的子序列长度l=2…k-1生成一个矩阵。然而,并非所有的矩阵都能提供相同的信息量。例如,给定长度的子序列的数目随着子序列的长度而减少,因此长的子序列提供的较少的信息量。此外,对于长代码,长度为2的子序列可能不会对PCR扩增产生影响。因此,合适的子序列长度是代码长度的25%,即最接近l=0.25*k的整数。对于固定的k,可以从矩阵Y生成一个热点图来推断指标较差或较好的子序列。此外,还可以对Y的元素进行聚合,以识别产生类似扩增指标的子序列的组成和位置。该方法可以采用在本文所述的癌症热点多重PCR分析中的商业化正常女性DNA(NormalFemaleDNA)模板上的几个样本的实验数据来说明,其中,随机8-mers被合成到一个目标扩增子的正向引物和反向引物上。代码引物既作为引物混合物的一部分,也单独作为单重PCR(singleplexPCR)使用。输入的DNA可以为m=500,1000,5000,10000,50000或100000单倍体基因组。分离的多重PCR反应进行15和25个循环。所有实验均在IlluminaMiseq平台上进行。表2显示了从每个样本中观测到的代码中所有可能的2-mers的频率排序。最有利的2-mers是‘AA’,而最不利的是具有高GC含量的那些,如'GG'或'CG'。表2.不同实验条件下24个样本按2-mers中位频率的排序列表。在Lasso方法中,可以将序列和热力学性质结合起来,以确定最有影响力的序列和热力学性质。该方法可以在用于本文所述的癌症热点多重PCR分析中的商业化正常女性DNA(NormalFemaleDNA)模板上来说明,其中,8-mers被合成到一个目标位点的正向引物和反向引物上。我们使用几个来自不同PCR循环(c=15,10,25和30)和输入量(m=7,575,0,500,1K,5K,10K,100K)的实验数据。所有实验均在IlluminaMiseq平台上进行。所考虑的序列的性质是子序列的位置和组成,其中子序列的长度为3。GC含量作为热力学属性被包括在内。采用三重交叉验证法(3-foldcrossvalidation)来确定调谐参数(tuningparameter)λ的最优值。表3列出了使用该调谐参数的Lasso方法的结果。该表格表明了与3-mers的子序列的位置和组成相比,GC含量对代码性能的影响更大。表3.具有GC含量和子序列的位置和组成的特征的Lasso方法中的系数基于设计标准为合适的代码搜索序列空间的随机迭代算法改进测量的或计算的参数可以用于设计约束条件来设计更大的最优性能的DNA代码池。在一些实施方式中,可以使用随机局部搜索算法((stochasticlocalsearchalgorithms)SLS)。例如,Tulpan等(10)所描述的SLS算法在具有固定大小的代码集合的空间执行局部搜索,这违反了给定的约束条件。这些约束条件可以包括本文所述的代码属性,也可包括涉及与池中其他代码的交互作用的约束条件,例如代码错配(C1)。搜索由随机选择的一组DNA链进行初始化。然后重复冲突,也就是,通过如下所述方法,选择一对违反约束条件的代码,并对各代码进行改造以解决冲突。输入参数约束参数C的列表,例如:n池的大小k字符长度dw池中字符对之间的代码间距ΔGheterodimer异二聚体形成的自由能阈值cGC含量该算法的参数如下:max_tries最大池初始化次数max_steps最大迭代次数nhood_size邻域大小(neighbourhoodsize)初始化随机选择一个初始字符集S,以满足GC含量的约束条件。可以使用[40%,60%]的GC含量来避免使用具有高的和低的扩增速率的代码。为了提高算法的性能,对满足GC含量约束条件的代码空间进行搜索。请注意,GC含量为c(其中40%<c<60%),长度为k的代码的总数为2k*∑j=[k*0.40]..[k*0.6]C(k,j),其中C是长度为k的代码中j位置的组合。然而,初始集通常包含一个较小的、满足GC含量约束条件的代码集合n。在整个算法中,集合的大小保持不变,并且在每次迭代中,尝试增加集合中满足约束条件的代码数量。邻域在每次迭代中,随机一致地选取违反约束条件的一对字符w1,w2∈S。然后建立w1和w2的邻域M,即,M=N(w1)UN(w2),其中N是由单突变邻域和随机邻域组成的混合随机邻域。给定代码w的单突变邻域由通过改造一个碱基从w获得的所有代码组成。对于给定的长度为k的一对代码w1和w2,有2*的单突变邻域满足GC含量约束条件。通过选择长度为k和GC含量为c的固定数量的随机代码来建立随机邻域。请注意,生成的随机代码的数量为nhood_size-2*k。随机邻域有助于避开搜索空间中的局部最小值。选择标准在邻域M=N(w1)UN(w2)中选择一个字符w',从而最大限度地减少在池中违反约束的次数。若w'∈N(w1)在池S中,则用S代替w1,或若w'∈N(w2)在池S中,则用w'代替w2,从而形成池请注意,池S和在一个字符中是不同的。停止标准在算法的每次迭代中,通过替换一个字符来修改池S。该过程执行最大max_steps次数。如果在max_steps迭代后未找到解决方案,则会随机初始化池S并重复该过程。池S最多初始化max_tries。当池S中的所有字符都满足约束条件或执行max_tries的最大值时,SLS将停止。图4、步骤(5)算法的伪代码(pseudocode)如下:程序随机局部搜索用于DNA代码设计请注意,在每次迭代中,会存储找到的最佳池Sbest,即违反约束条件数量最少的池。SLS返回Sbest。另外,请注意,该算法有两个循环语句(forloops)。在外部循环语句中,池被初始化,因此代码的实现可以与SLS的max_tries独立运行并行。应该理解的是,可以使用本文所述的SLS的修改的版本,或另一优化方法来找到满足一系列约束的池。通过代码熵来分析模板的多样性在某些方面,本公开提供了在源自模板分子池的扩增序列、质量控制、突变识别以及其他用于NGS测序的应用中,利用信息理论测定代码熵的方法。在一些实施方式中,代代码是连接的(例如,与目标引物序列连接(ligated)或一起合成,如本文所述的那些用于CG001.v2的引物序列)。代码与引物的连接,通常可以绕开连接到模板分子上的低效率和不可预测性,而这对于从档案样本中重新获得的DNA模板(例如用于患者组织常规诊断方法的福尔马林固定石蜡包埋(FFPE)组织样本)来说尤其成问题。在可替代的实施方式中,代码可以连接到目标分子上。因此,在一些实施方式中,将本文所描述的方法可以应用于模板-代码连接的模板上。由于NGS过程中使用了一对末端测序方法,因此在NGS测序反应、分子条形码和引物中启动了两个不同的引物,见图5。在一些实施方式中,接头可用于识别为每个末端设计的样本。此外,一个不同的条形码被连接(例如通过连接方式或合成途径)到每个目标引物,以提高编码效率。获得的经修饰的引物还可以通过,例如额外的PCR反应(其中样本通过额外的DNA索引进行编码),进一步包括用于在多路分解步骤(demultiplexingstep)中连接的通用接头序列。然而,对模板多样性的分析并不需要使用接头序列。在一些实施方式中,将单个代码或分子条形码连接到引物对中的两个引物其中之一上。在可替代的实施方式中,在引物对中使用相同或不同的代码或分子条形码。在可替代的实施方式中,与接头序列连接或不连接的代码或分子条形码可直接连接到核酸模板分子(例如DNA模板分子)上,以形成代码-模板分子。并且随后生成的嵌合的模板-代码[-接头序列]分子可使用通用引物进行扩增。不受任何特定理论的束缚,该方法可用于对来自全基因组的DNA片段池,或来自对基因组DNA片段的杂交的富集捕获中的DNA片段池进行测序。在这种情况下,本领域技术人员能够应用本文所描述的熵的方法。总之,通过代码熵分析模板的多样性,可通过以下步骤进行:●采用均衡的或不均衡的性能的代码库,将代码随机连接到扩增产物或模板上;●使用例如CG001v2分析中所描述的方法对目标位点进行PCR-NGS测序;●代码丰度的校准和计算;以及●将观测到的熵与预期的熵进行比较,并结合判定程序通过模板池大小的伪差(artifact)和估值来确定真正的变异。在PCR测序过程中,采用性能理想的代码池估测DNA代码的预期的熵对不同性能特征的代码熵的期望测定方法可以按如下方式确定。可以在接下来的步骤中采用期望的方法来确定实际的性能并识别突变。在一些实施方式中,生成一组多元性高的代码M的池,并通过例如合成的或连接的方式将其连接到目标引物上。在可替代的实施方式中,通过例如连接的方式将DNA代码可随机地连接到模板分子上。在一些实施方式中,所观测到的代码的多样性可以使用Shannon熵来确定。然而,应当理解的是,可以使用任何其他合适的多样性度量标准,如辛普森指数。PCR反应开始于初始数目的模板分子m,其将与代码连接的引物池交互作用。采用Shannon熵H来定义在c循环数(Ac)的PCR程序中扩增的产物中观测到的给定代码集的多样性,Shannon熵H定义为其中当P(wj)=0时,P(wj)log2P(wj)=0。因此,在给定的PCR循环中所观测到的代码的熵取决于几个因素,如池的大小||,代码池M中每个代码wj的重数i(wj),以及模板分子m的初始数目。在给定的PCR循环中代码的熵可按如下方法来估算。首先,估算每个PCR循环中所需的扩增序列的数量和unique代码的最小数。生成两个不同的代码池,一个用于正向引物MF,另一个用于反向引物MR。因此,在给定的PCR循环结束时,观测到与扩增产物相关的两组代码:一组用于正向引物,另一组用于反向引物。例如,图6显示了前4个PCR循环的扩增序列以及与每个扩增序列相关的正向引物和反向引物的代码。表4包含了在perfectPCR过程的每个循环中发现的代码的列表以及相应的熵。例如,在第4个PCR循环中,有22个扩增序列,在每个末端有14个unique代码,一个代码熵为3.66。表4对perfectPCR反应的前4个循环中观测到的扩增序列和代码的数量的统计一般可以推断出扩增序列的数目和unique代码数。在给定的PCR循环中,可以有三种类型的序列:(1)初始的DNA模板,(2)来自一端有一个代码的初始模板的引物延伸产物,(3)来自含两个代码(一端有一个代码)的引物延伸产物的引物延伸产物。表5包含了在给定的PCR循环中观测到的每种类型的序列数。它还包含一个通式,用于获得在任意给定的PCR循环c中观测到的每种类型的序列数。表5.每个PCR循环中发现的不同类型的序列数。为获得每个PCR循环中unique代码的数量,请注意,来自初始DNA模板的每条引物延伸产物都包含一个代码w1。然而,当扩增时,产物将包含代码w1和在另一端的新代码w2。类似地,来自引物延伸产物的引物延伸产物包含两个代码w1和w2。这些序列将在一个方向进行扩增,新产物将包含一个新的代码w3。因此,每当一个序列被扩增时,都会引入一个新的代码(图7)。由于每种类型序列会在下一个循环中产生新的代码,因此每个循环c的unique代码总数等于2c-2(表6)。表6.每个PCR循环的unique代码的数目在perfectPCR过程中,给定的代码Wj的频率fi,c在循环数c时可以计算为fi,c=fi,c-1+1,时,c0为wj首次被观测到的循环数。也就是说,在每个PCR循环中,代码频率预计会增加一次。表1显示了图6中第一个和第二个循环中出现的代码的频率。理想情况下,代码池中每个代码都是均匀分布的,且重数为1,即i(wj)=i~Uniform(1),其中Uniform是指均匀统计分布(Uniformstatisticaldistribution)。然而,实际上,由于寡核苷酸合成中的错误,对序列固有的热力学约束导致的某些序列寡核苷酸合成的低效率,低效率的PCR扩增,以及由于类似问题而导致的代码排序等因素,所观测到的多重数分布可能不同于所述的均匀性。事实上,任何编码方法都可能受到非均匀特性的影响。代码非均匀分布的影响可按如下方法处理,以提供对PCR测序过程中熵特征的估算。第一步是识别代码多重数的经验分布。理想的分布是均匀分布,用来对一段时间内观测到的事件的数量进行建模,但是实际上也可以观测到其他类型的分布,例如泊松分布。当分布的均值和方差不同时,也可以观测到负二项分布。作为第一步,可通过探索性分析和Q-Q统计图对经验分布和已知分布进行比较。然后,在给定所选概率分布模型的情况下,可以使用最大似然估计来获得观测的代码多重数分布的概率。此外,拟合优度检验也可以用来指示假定随机样本来自特定分布是否合理。下一步是在给定已经表征的特定代码多重数分布的前提下,确定预期的代码熵分布。在给定的PCR循环中,观测到的代码可以采用在代码池||中统计回置抽样法(statisticalsamplingwithreplacement)来建模,其中样本的大小取决于扩增序列的数目。由于根元素wj的重数可以大于1,因此可以采用回置抽样法。此外,PCR反应过程中的错误会影响代码的熵,例如,引物可能会解离并重新启动。以下几部分内容描述了当存在不同的多重数代码分布时,在第4个PCR循环中代码熵的行为。在每种情况下,通过对确定根元素的多重数分布的固定池生成1000个独立放回抽样,来得到熵的分布。按以下所述方法,通过模板数在m=1(模板数定义为m)和m的倍数之间的代码的熵的行为,来说明熵方法如何区别在真正的等位基因的PCR测序过程的后期循环中引入的错误,或者随机分布的单模板变异。(i)均匀多重数分布(UniformMultiplicityDistribution)在优选的实施方式中,包含4个循环的perfectPCR反应有14个不同的代码(见图6,表4)。如果i(wj)=i~Uniform(1),数量为m的模板分子扩增所需池的大小为|M|=m*∑j∈|W|i(wj)=m*14。这相当于回置抽样的群体大小。由于每个模板分子在4个PCR循环后具有22个扩增序列,因此样本大小为n=22*m。在这种情况下,在池中观测到给定代码的概率是P(wj)=i(wj)/|M|=1/(m*14)。图8显示了具有不同初始模板数m的观测到的熵的分布。这些分布是通过计算来自参数为1的均匀的代码多重数分布的1000个独立回置抽样的熵产生的。当初始模板分子数m增加时,会观测到更大的熵。然而,熵的方差随着m的增加而减小。图8还包含了对于不同的m值,用水平线表示的预期的熵。预期的熵总是高于观测到的熵。当代码的多重数均匀分布时,熵的分布与代码的多重数i(wj)无关。这在图9中用i~Uniform(1)和i~Uniform(3)表示。理想情况下,代码的多重数是均匀分布的。然而,由于寡核苷酸合成中的错误,多重数可能发生变化。然而,当池中为不均衡的代码时,本文所描述的熵方法仍然可以用来区分在后期的PCR循环中出现的错误。(ii)泊松多重数分布代码多重数的变化可以使用具有参数λ的泊松分布来建模,即i~P(λ)。所述泊松分布用于对一段时间内观测到的事件数量进行建模。在这种情况下,事件是在寡核苷酸合成过程中生成的代码wj。如果i~P(λ),并非所有代码具有相同的多重数,然而平均值和方差等于λ。泊松分布的密度函数定义为其中μ[i]=λ=σ2,k∈{0,1,...}。为了对这一情况进行建模,生成了一个泊松样本,并且由于代码多重数i应该大于零,因此值移位1。图10显示了随机生成的泊松分布以及修正后的泊松分布,其中所有值都移位1。请注意,在该移位分布中,均值增加了一个单位,但方差保持不变。当循环数大于1时,PCR过程的质量可以更好地进行评估。这是因为如果模板在PCR循环c结束时没有得到很好的扩增,扩增后产物的代码的熵可以被识别为与低PCR循环c'有关的预期的熵,其中c'<c。在PCR测序过程中,用非均匀性能的代码池来估算DNA代码的预期的熵。图11和图12显示了当多重数服从均匀分布以及具有不同λ值的泊松分布时的熵的分布。通过计算来自固定的代码多重数分布的1000个独立样本的熵,来获得熵的分布。当多重数服从泊松分布时,熵会减小。原因在于一些代码的出现概率较高,因此,当所有代码都有相同的出现概率时,与所得到的样本相比,样本多样性会减小。图13显示了当多重数为均匀分布和参数λ=i=1的泊松分布时,对池中每个代码进行抽样的概率。当多重数遵循泊松分布时,随着池中代码出现的次数变得更均匀,熵随λ值的增大而增大。此外,随着初始模板分子数m的增加,熵增大,而方差减小。(iii)负二项多重数分布泊松分布是假定分布的均值和方差是相同的。然而,实际上当多重数的方差大于平均值时,可以观测到不平均分布。这种情况可以用负二项分布来建模,即i~NB(r;p)。该分布对在规定次数的失败r发生之前,在一系列独立的伯努利试验中成功次数发生的概率进行建模。每次伯努利试验的成功概率是p。密度函数定义为P(i=k)=C(k+r-1,k)pk(1-p)r且k=0,1,2,...,其中C是k+r-1伯努利试验中成功k的组合。平均值和方差分别为μ[i]=pr/(1-p)和σ2[i]=pr/(1-p)2。为了对这一情况进行建模,生成了一个负二项样本,并且由于代码的重数i应该大于零,因此值移位1。图14显示了随机生成的参数r=6和p=0.5的负二项分布,以及所有值都移位1时修正后的分布。请注意,平均值在移位分布中增加了一个单位,但方差保持不变。为了考察方差对熵的影响,用负二项分布的不同参数生成了几个样本。表7包含每个生成的样本的平均值和方差。参数p和r变化,以使样本平均值固定。例如,当时,方差的值范围为2.08至10.3。请注意,随着成功概率p的增加,样本方差(σ2)减小。此外,对于固定p,样本方差随样本平均值的增大而增大。表7不同参数的负二项分布样本的均值和方差。每个样本都用参数p和r=(1-p)/p生成,p=0.1,0.2,…,0.9,μ=1,3,6。请注意,因为样本的值移位1,为了对第4个PCR循环进行建模,其中DNA模板的初始数量3000,将每个样本的大小固定为42,000。图15显示了当多重数服从参数1的均匀分布和μ=1,3,6,p=0.1,0.2,...,0.9时的负二项分布时的熵的分布。通过计算给定代码多重数分布的1000个独立样本的熵,得到熵的分布。当代码多重数服从负二项分布时,所观测到的熵比均匀代码多重数分布时的熵低。对于固定p,熵随μ的增大(即当样本方差增大时)而减小。此外,对于固定的μ,熵随参数p的减小而减小,因此方差增大。图16显示了平均熵与每个分布的方差之间的关系。对于固定的样本均值,熵随多重数分布方差的增加而减小。图17A-C和18A-C显示了当模板分子的初始数为m=1,5和10时的代码熵。一般来说,熵随着方差的增加而降低。随着m的增加,这一趋势变得更加明显。(iv)具有异常值的均匀多重数分布实际上可能出现的另一种情况是,除少数代码具有较高或较低的出现次数外,大多数代码都具有相同的多重数。在这种情况下,多重数被建模为具有一些异常值的均匀分布。在具有4个循环和m个初始模板拷贝的PCR过程中,池的大小计算为每个代码的抽样概率为为了对这一情况进行建模,采用不同数量的异常值和范围在5至7之间的随机多重数来生成参数1的均匀分布。图19显示了当m=5且|W|=14*5=70时,来自1000个独立样本的相应的熵的分布。当引入异常值时,熵会下降。然而,对于少数异常值,可以观测到熵的较低值。然后熵随异常值数量的增加而增加。当异常值数量为70时,即在每个代码中都引入异常值时,所得到的熵与无异常值时得到的熵是具有可比性的。(v)均匀多重数分布和不同PCR循环数实际上,并不是所有的序列都像预期的那样进行扩增。例如,某些序列只在PCR过程的早期循环中进行扩增。为了对这一情况进行建模,当多重数为均匀分布时,我们比较了在不同PCR循环数中扩增的序列的代码熵。对每种情况进行建模所需的参数是在不同PCR循环数的群体大小、样本大小和代码采样的概率。这些参数包含在表8中。表8用于模拟2个,3个和4个不同循环的PCR扩增的参数。这些参数对应于多重数为均匀分布的情况。图20显示了当i~U(1)时不同PCR循环的熵的分布。使用表8中所示的不同PCR循环的参数来生成1000个回置抽样,从而得到熵分布。在PCR循环较少的模拟中观测到的熵值较低。因为可以预期到较少的循环具有较少数量的unique代码。代码引入对扩增子性能的影响采用商业化的正常女性DNA模板来检测扩增子性能,其中10-mers合成到我们的癌症热点多重PCR分析中的所有73个目标位点的正向引物和反向引物上。我们使用了25个PCR循环和不同数量的输入DNA(m)。当m=5,000和10,000时,将来自该实验的每个扩增子的读数数量与包含商业化的正常女性DNA模板和不含代码的引物的4个不同实验进行比较。这些实验是在IlluminaMiseq平台上进行的。在这4个实验中,我们使用了30个PCR循环和m=7575单倍体基因组。图23A和B表明,尽管PCR循环较小,但有代码的性能与没有代码的性能相当。请注意,在确认池中没有优先扩增的代码可能对结果产生偏差后,才能分析扩增子的性能。起始模板与熵的关系z作为模板初始数的函数的熵预期会增加。这一关系的一个例证就是商业化的正常女性DNA(Coriell生物库)模板,其中随机8-mers和10-mers被合成到我们定制的CG001癌症热点多重PCR分析的一个目标扩增子的正向引物和反向引物上。使用MiSeq平台对读取进行抽样。对于8-mers的实验条件为:20个PCR循环和m=10,50,100,500,1000,5000,10000,50000,100000的DNA的输入量。对于10-mers的实验条件为:25个PCR循环和m=1,2,3,4,5,10,25,50,75,100,500,1000,2000,3000,4000,5000,10000,25000的DNA的输入量。图24A和B分别显示了来自5号染色体位置136633338的8-mers和10-mers的SNPrs13182883的每个等位基因的代码熵。这些图将熵表示为初始模板数m的函数。对于SNP等位基因A和G,当输入DNA的范围大约为m∈[10,4000]时,代码熵总的趋势是增加的。随后每个扩增子的代码熵按输入DNA的函数进行分析。图25显示了几个起始模板的代码的熵的分布情况。所有代码的熵是根据来自同一扩增子的读数计算出来的。图25显示了当50≤m≤4000时预期的趋势,其中中位熵随初始模板数的增加而增加。用于质量保证的DNA条形码的应用诊断DNA测序中的质量保证可以避免对患者的治疗和管理提供错误信息。在NGS方法中,非常需要和能够用于质量保证的不同方面(从过程污染的检测、样本一致性,到分析有效性的精确定义)的方法一起使用。DNA代码可用于评估背景中介绍的目标测序范例中扩增产物的不同方面,并用于以下各个目的。(1)检测样本或过程污染选择按本文所述的方法生成的不同组的含非重叠成员属性的已知代码池,用于不同日期的处理,或用不同批次样本处理,例如通过引入CG001v2的引物序列,但任何其他能够靶向基因组区域的引物序列也可以使用。因此,每个实验随时都有一个不同的代码集在使用。在一些实施方式中,代码连接到靶向人类基因组中已知的多态单核苷酸变体的引物上。使用大量个体的生殖系多态性,通过检测到的多态变体的组合来区分不同的人个体。后者可包括单碱基变异、缺失或重复序列的变异。所选择的多态数可以根据群体中某一给定多态性的频率和位点数来确定一个函数,从而将偶然双重发生的可能性降低到可接受的阈值以下。可接受的阈值可以是1/1000000,也可以使用1/1000至1/1000000之间的或小于1/10000000的任意值。合适的单碱基多态性的数量可以是16个,但也可以使用10,11,12,13,14,15,17,18,19,20,21,22,23,24,25,26,27,28,29,30或更多个。生殖系多态性和DNA条形码的双重使用使得在多重测序和信息学实验步骤中,能够唯一地识别单个DNA模板,并且定义的代码集的存在,可以允许对实验室工作流程中的板与板之间,分析与分析之间或日间的交叉污染进行检测。(2)检测PCR中模板多样性不足的代码多样性代码熵的分布的实际性能可以按照如本文所述的方法,从在独立DNA模板控制下的几个连续稀释试验获得,所述试验包括将约3000个拷贝的模板逐步稀释至单个拷贝,并对已知模板在不同稀释度时不同目标位点的熵进行测定。在一些实施方式中,可以使用少至4个分子。在可替代的实施方式中,可以使用多于3000个拷贝。在可选的实施方式中,可以使用4至3000个拷贝或它们之间的任意值,例如10,50,100,500,1000,1500,2000,2500等。连续稀释可获得不同浓度的初始模板分子。本领域技术人员熟悉如何以类似于在一系列测量中使用确定的输入来使分析性能标准化的任何分析的方式,来进行连续稀释实验,以获得作为输入的起始模板与作为输出的熵(该方法的性能测定)之间的关系。更高浓度的初始模板分子预期会有更高的代码熵。对此在图26中以等位基因SNPs的熵进行举例说明。对于固定的浓度,实验至少进行一次,但最好重复两次或多次来获得代码熵的分布。然后,对于感兴趣的DNA模板,将具有相同浓度初始模板分子的扩增产物的熵与相应的预期的熵的分布进行比较。如果相关的熵的测定值比预期的值小,那么反应可能会因为不充分而被拒绝。这些信息被纳入整个样本的处理过程。在PCR反应中,质量保证还将纳入对不同年龄和性能的模板的参考测量,并在不同天进行重复,作为全过程保证的一部分。当扩增产物低于预期值时,观测到的熵低于预期的熵的分布,见图21A和图21B。因此,在经验的熵分布中观测到的H(扩增产物)的概率接近于零。如果经验的预期的熵分布呈参数为μ和σ2的正态分布,则可以使用Z-评分测试来确定扩增产物的熵x=H(扩增产物)是否位于预期的熵的分布的尾部。给定值x的Z-得分定义为且是偏离平均值的标准差的测量。在Z-评分测试中,零假设定义为Ho:x=μ。如果p值小于显著性水平α,则零假设被拒绝。在正态分布的尾部发现与非常小的p值有关的非常高或非常低的(负)Z得分。这表明观测的值x不太不可能的属于预期分布N(μ,σ2)。除Z-评分方法外,其他方法也可以应用。例如,在假设观测到的熵属于预期的熵的分布的前提下,可以确定观测到的熵的分位数。如果观测到的熵是一个离群点,那么这表明PCR过程中有一个伪差,并且允许在测序/质量控制过程中对样本的拒绝。模板分子中真正的突变的检测,与PCR/测序错误或随机分布的单个碱基变异本文所述的一种或多种方法可用于例如癌症诊断,其中恶性细胞亚群可包含大多数人中不存在的变体(称为克隆)。在病原体测序领域的其他应用中,可以检测到群体中罕见的细菌或病毒基因组。本文所述的一种或多种方法通常可用于任何情况,其中,在人口背景中通过NGS测序对罕见的DNA变体(“低发的真正的突变”)进行分析/检测。应当理解的是,本文所描述的方法可以用于具有任意变体等位基因发生率的任何序列,而且不需要这种变体是一种罕见的变体。这些方法在假设变异等位基因的代码熵的分布和背景不同的情况下进行。这可以通过将与SNPs相关的等位基因和因测序错误或伪差引起的低频等位基因的代码熵进行比较来说明。表9列出了在正常女性样本中发现的SNPs。该分析所考虑的伪差位置(具有测序错误的位置)位于表10所列SNPs的邻域[SNP-5,SNP-3]和[SNP+3,SNP+5]。在伪差类型(artifactclass)中,代码熵是根据次要SNP等位基因和所有低发的等位基因计算的,参见图26和27。而当25≤m≤4000时,含有伪突变的等位基因的中位熵(medianentropy)保持不变,而SNP等位基因的中位熵增加。表9显示了在每个连续稀释的正常女性样本中确定的SNPs。通过几个实验,在本文所述的肿瘤热点多重PCR分析中使用商业化的正常女性模板,证实了SNPs和等位基因SNPs,实验条件为:30个PCR循环,m=7575,不含代码的引物。次要等位基因和该表中所记录的%VAF与含有代码的实验(25个PCR循环,不同数目的初始模板)相当。表10显示了考虑的伪差的位置是该表所列的SNP位置的邻域[SNP-5,SNP-3]和[SNP+3,SNP+5]。表10SNP染色体位置rs6811238chr4169663615rs576261chr1939559807rs10092491chr828411072rs1821380chr1539313402rs9951171chr189749879rs1058083chr13100038233rs13182883chr5136633338rs2981448chr10123279745rs2071616chr10123279795rs3738868chr229432625rs1136201chr1737879588rs1050171chr755249063rs12628chr11534242rs2230587chr165311262rs2228230chr455152040使用监督或非监督分类方法,可以将低发的真正的变体与测序错误区分开来。监督分类方法是本领域技术人员已知的,包括但不限于使用训练集的方法。考虑的类型是(1)真正的突变和(2)测序和/或标记为伪差的聚合错误。分类方法的性能取决于选定的特征。我们通过以下方法来证明几种采用两个特征对变体进行分类的监督方法的性能:(1)低发的变体的扩增读数的代码熵;以及(2)覆盖范围定义为在该变体位置上的扩增读数的数量。除非特别指定,使用来自python的scipy库来运行含默认参数的算法。-具有平衡权重的线性支持向量机(SVM),其中与分类相关的权重与分类频率成反比。即,其中y∈{artifact,mutation}。-具有平衡权重的径向基函数(RBF)SVM。-最近邻。通过分配最接近查询点的k个训练样本中出现最频繁的标签,来对测试点进行分类,其中k=3。-具有平衡权重的逻辑回归。-AdaBoost算法-线性判别分析-随机森林算法,其具有max_depth=5的树的最大深度,在查看最佳分割max_features=1时需考虑的特征数以及平衡权重。-二次判别分析-决策树,其具有max_depth=5的树的最大深度以及平衡权重。-高斯朴素贝叶斯模型采用正常女性基因组DNA和HorizonQMRS多重参照DNA(从FFPE卷轴自身制备(preppedin-housefromFFPEscrolls))的混合物,通过将随机10-mers合成到全部73个CG001目标位点的正向引物和反向引物上,来对这些方法进行测试。PCR反应进行25个循环。每个反应DNA的组合输入保持在5000单倍体拷贝,以及两种混合物:(1)100%QMRS和(2)10%QMRS+90%正常女性基因组DNA。表11显示在真正的突变类型中所考虑的突变的列表(在与NormalFemale(NF)相结合的QMRS中发现的突变的列表)观测到的真正变异的变异等位基因频率(VAF)的百分比介于0.86%和25.62%之间。从所有与真正的突变位置不同的几个位置处的低发的等位基因的实验中获得伪差类型的数据。考虑的位置是表10中所列的SNPs的邻域[SNP-10,SNP-5]和[SNP+5,SNP+10],以及表12所列的外显子区域。正常女性样本的系列稀释的伪差位置[SNP-5,SNP-3]和[SNP+3,SNP+5]也包括在内。图28示出了所有数据的熵和覆盖范围,其中指明了伪差类型,以及具有相应变异等位基因频率百分比的真正的突变类型。此外,训练数据和测试数据也被标记在相同的图中。表13示出了预测的测试集中真正的突变类型。图28测试集中的突变数据以及来自每个分类器的预测的类型也都包括在内。该测试集的性能指标以马修斯相关系数表示。请注意,马修斯相关系数考虑了所有测试数据,而不仅仅是该表中显示的突变测试数据。通过20折交叉验证获得每种分类方法的性能。使用交叉验证的分层策略来确保每一折大致包含相同比例的两个类型。由于伪差类型的尺寸比真正的突变类型大得多,定义为MCC=(TP*TN-FP*FN)/[(TP+FP)(TP+FN)(TN+FP)(TN+FN)]1/2的马修斯相关系数被用作性能指标。表13.监督分类方法的性能表14表明非概率方法SVM和最近邻以及在本研究中性能表现最好的逻辑回归概率方法。将分层交叉验证运行20多次的马修斯相关系数的平均值作为性能指标。因此,在一些实施方式中,在本文所描述的方法中使用的监督分类方法包括马修斯相关系数至少为0.7的方法。这些方法包括但不限于SVM,最近邻和逻辑回归概率方法。表14监督分类方法的性能在NGS测序中引入基于对模板复杂度和核苷酸变异的测定的熵图22中所概述的过程示出了如何在实际中使用所观测到的代码熵来检测扩增产物的质量。该过程首先表征所有观测到的代码。用于检测先前实验中的污染的代码的存在表明了污染的存在。如果没有污染,这个过程将继续,并用一种方法来检测测序过程中模板的用量不足。如果序列像预期的那样被扩增,那么最后一步就是检测真正的变体。图22中所示的程序在初始假设代码多重数的分布是均匀的前提下进行。由于技术问题引起的代码熵的不均匀性可以按本文所述的方法来检测,并且因此并入到预期的背景熵的计算中。用NGS测序panel对患者肿瘤组织进行测序的样本工作流程。提出请求的医生将访问一个安全的外部门户网站,提交病人样本的申请表。样本一经收到将被录入公司的实验室信息管理系统(LIMS),并且针对患者的用福尔马林固定石蜡包埋的组织的肿瘤细胞结构,用苏木精和曙红(H&E)载玻片进行评估。如果病人的样本没有足够的肿瘤含量,将需要一个新的样本。如果样本在提取后不能产生超过100ng的DNA,则还需要新的样本。在库建设和数据分析后,样本还需要满足所有的QC要求。一旦所有的QC指标都通过了,将生成病人报告,并分发回提出要求的医疗服务提供者。图1显示了患者样本工作流程。DNA提取用QIAampDNAFFPE组织试剂盒(Qiagen)从4x10微米的福尔马林固定石蜡包埋的组织中提取DNA。对提取方案进行修改以便脱蜡,包括将样本在矿物油中加热至90℃。简言之,将300ul分子级矿物油加入到FFPE卷轴中,并在90℃加热20分钟。在加入ATL缓冲液和蛋白酶K后,严格按照Qiagen’s的说明进行处理样本。为了有助于从熔化的石蜡中分离水层,在将液体转移到旋转的柱上之前,将样本在冰上冷却4分钟。用QubitFluorometer(LifeTechnologies公司的Invitrogen)来定量检测洗脱的DNA。文库构建采用50ngFFPEDNA,用QiagenMultiplexPCR试剂盒生成扩增子。这些扩增子在两个池中产生:池A和池B,共有73个扩增子(表15所列引物),涵盖90多个热点和7个外显子(表16)。表16.扩增子涵盖的热点和外显子。位点特异性引物包括NexteraXT(Illumina)常见序列,以便在PCRAmpureXP磁珠清理之后,使用NexteraXT条形码试剂盒进行文库构建。通过8个PCR循环将编入索引的接头连接到扩增的序列上。当文库构建样本再次用AMPureXP磁珠进行纯化后,用Qubit进行定量,用安捷伦生物分析仪(AgilentBioanalyzer)进行分析。将样本合并并稀释至12.5pM,然后使用300个循环v2试剂盒对配对的末端150bp读长在MiSeq(Illumina)上进行测序。合并的策略是这样的,每次运行包括20名患者、一个阳性对照和一个阴性对照。癌症患者中靶向扩增体细胞畸变的引物PanelCG001.2用primer3(3)设计包含73个PCR引物对的目标寡核苷酸DNA序列引物panelCG001.v2(表15),来扩增人类基因组中的目标基因区域(hg19)。用于引物设计的基因组区域包括目标区域的上游和下游200bp的基因组区域。对在panel中使用的引物对的选择涉及使用primer3对每个目标区域涉及包含至少45个引物对的引物组的设计,primer3的设置包括:最小大小:18,最佳大小:20,最大大小:27,产物大小范围:100-249,最低温度:57,最佳温度:60,最高温度:63。符合下列任何一项标准的引物对将被排除在设计组之外:在正向引物或反向引物序列中有多于三个连续的鸟嘌呤,引物比对到含1000个基因组等位基因snps的发生率>0.005的基因组目标区域,使用NCBIBlast(4)确定的偏离目标基因组区域扩增的引物对。依次测试每个引物组中的引物对与两个池中的一个池中的已有引物对的兼容性。测试每个引物对与已有引物对的二聚作用,定义为>80%碱基匹配时有大于4个碱基对齐。一旦识别出兼容的引物对,就会将引物对添加到池中,并终止对引物组的引物对测试。然后开始对下一个引物组中的引物对进行测试。使用此过程创建的最终引物panel包括从被拆分到两个池中的每个引物组中选择的兼容的引物对。评估每个panel中每个引物对的PCR扩增性能,并使用primer3重新设计未能扩增基因组序列的引物对,并测试其与已有池的兼容性。来自执行CG001.v2的序列的信息分析采用bwa和BWA-mem算法(5),使由IlluminaMiSeq产生的目标扩增子的配对读长可以与参考基因组hg19比对。使用SAMtools(6)和bamUtils(7)对比对读长进一步处理和过滤。只有符合以下标准的比对读长才用于进一步分析;在预期读长的目标序列上,少于5个错配的读长,以及softclipping小于7bp的读长。然后分别采用MutationSeq(2)(http://compbio.bccrc.ca/software/mutationseq/)和strelka(8)工具,使用过滤后的比对来识别SNVs和indel。MutationSeq使用基于特征的分类器来评估在任何给定位置发生体细胞突变的概率,并需要匹配的肿瘤-正常对的测序数据。Strelka基于贝叶斯方法,并且也需要肿瘤-正常对。由于不存在匹配的正常样本,因此使用来自健康女性个体的正常B-淋巴细胞的细胞系作为正常参照物,来对变体进行检测(NA01953,CoriellBiorepositories)。对置信度高的SNVs的检测要求目标最小深度为1000x,MutationSeq概率得分>=0.9。插入缺失的检测要求最小目标深度为1000x,质量评分至少为30。在较宽的等位基因频率(1~33.5%)范围内,使用QuantitativeMultiplexDNAReferenceStandard(HorizonDiagnostics)作为检测SNVs和插入缺失的阳性对照。预测的等位基因频率相对于已报道的阳性对照的等位基因频率的皮尔逊相关系数至少为0.9,其作为是成功检测到变体的标志。使用SnpEff(9)和UCSC已知基因数据库对检测到的高置信度SNVs和插入缺失的影响进行注释。分析工作流程如图2所示,其进一步的扩展包括图22所示的工作流程,其包含了从代码分析到突变识别的已公开的方法。参考文献1)Goya,R.,Sun,M.G.,Morin,R.D.,GG,L.,Ha,G.,Wiegand,K.C.,etal.(2010).SNVMix:predictingsinglenucleotidevariantsfromnext-generationsequencingoftumors.Bioinformatics(Oxford,England),26(6),730–736.http://doi.org/10.1093/bioinformatics/btq040)2)DingJ1,BashashatiA,RothA,OloumiA,TseK,ZengT,HaffariG,HirstM,MarraMA,CondonA,AparicioS,ShahSP.Feature-basedclassifiersforsomaticmutationdetectionintumour-normalpairedsequencingdata.Bioinformatics.2012Jan15;28(2):167-75.http://doi.org/10.1093/bioinformatics/btr6293)UntergasserA,CutcutacheI,KoressaarT,YeJ,FairclothBC,RemmMandRozenSG.(2012),Primer3--newcapabilitiesandinterfaces.NucleicAcidsRes.40(15):e115.4)StephenF.Altschul,ThomasL.Madden,AlejandroA.JinghuiZhang,ZhengZhang,WebbMiller,andDavidJ.Lipman(1997),"GappedBLASTandPSI-BLAST:anewgenerationofproteindatabasesearchprograms",NucleicAcidsRes.25:3389-3402.5)LiH.Aligningsequencereads,clonesequencesandassemblycontigswithBWA-MEM.(2013).arXiv:1303.3997v1[q-bio.GN]6)LiH1,HandsakerB,WysokerA,FennellT,RuanJ,HomerN,MarthG,AbecasisG,DurbinR;1000GenomeProjectDataProcessingSubgroup.TheSequenceAlignment/MapformatandSAMtools.Bioinformatics.2009Aug15;25(16):2078-9.7)BreeseMR1,LiuY.NGSUtils:asoftwaresuiteforanalyzingandmanipulatingnext-generationsequencingdatasets.Bioinformatics.2013Feb15;29(4):494-6.8)SaundersCT1,WongWS,SwamyS,BecqJ,MurrayLJ,CheethamRK.Strelka:accuratesomaticsmall-variantcallingfromsequencedtumor-normalsamplepairs.Bioinformatics.2012Jul15;28(14):1811-7.9)CingolaniP1,PlattsA,WangleL,CoonM,NguyenT,WangL,LandSJ,LuX,RudenDM.Aprogramforannotatingandpredictingtheeffectsofsinglenucleotidepolymorphisms,SnpEff:SNPsinthegenomeofDrosophilamelanogasterstrainw1118;iso-2;iso-3.Fly(Austin).2012Apr-Jun;6(2):80-92.10)TulpanDCandHoosHH(2003).HybridrandomizedneighbourhoodsimprovestochasticlocalsearchforDNAcodedesign.LectureNotesinComputerScience2671:418:433.所有引文均以参考方式纳入。本发明描述了一个或多个实施方式。然而,对本领域技术人员显而易见的是,在不脱离如权利要求中限定的本发明的范围的情况下可以进行多种变化和修改。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1