双二甘酰胺配体及其制备方法和含有双二甘酰胺配体的镧系/锕系分离萃取体系与流程
本发明属于核燃料循环及核废物处理技术领域,涉及用于镧系/锕系分离的萃取体系,具体涉及一种双二甘酰胺配体及其制备方法和含有双二甘酰胺配体的镧系/锕系分离的萃取体系。
背景技术:
核燃料的合理处理、处置是核能可持续发展的关键问题之一。“分离-嬗变”法是一种普遍优先采用的核燃料后处理方法,其要点是采用溶剂萃取法从高放废液中将占例较小但毒性极强的锕系选择性分离出来,然后将其嬗变为短寿命或稳定的核素。目前,较成熟的用于锕系分离的流程主要有TRUEX、DIDPA、TRPO、DIAMEX等流程(顾忠茂等,原子能科学技术,2002,36(2):160-167),但这些流程在萃取锕系的同时亦会将镧系一起萃取,而镧系有较大的中子吸收截面,会成为锕系嬗变中的“中子毒物”,严重干扰其嬗变,甚至无法进行。因此,镧系/锕系分离在高放废液处理中是十分必要的。然而,由于三价锕系与镧系具有非常相近的物理和化学性质,因此分离难度极大。尤其是锕系中需要处理的最关键元素为镅(Am),而与其物理化学性质最相近的镧系是铕(Eu),若能实现Am/Eu的分离,则镧系/锕系分离问题可迎刃而解。因此,目前该领域的工作主要以Am和Eu作为锕系和镧系的代表进行研究。
依据软硬酸理论,虽然三价锕系与镧都属硬酸,但前者比后者略偏软一些,更易与含S、含N等软配位原子的配体结合,理论上可利用这一差异实现镧系/锕系的分离。因此,含S、N等软配位原子的萃取剂的研发成为多年来该领域的研究重点。
目前国际上普遍采用的镧锕分离方案,是先将被共萃的锕系与镧系用0.1~0.5mol/L的HNO3反萃,再从反萃液中将锕系或镧系分离出来(Madic C et al.,PARTNEW—New solvent extraction processes for minor actinides—Final Report;2004;CEA-Report 6066)。从上世纪60年代开始,国际上开发了许多用于镧锕分离的流程,代表性有TALSPEAK、Cyanex 301、BTPs等流程。
TALSPEAK流程是由美国橡树岭国家实验室于1964年提出的(Nilsson M et al.,Solvent Extr.Ion Exch.,2009,27:354-377)。该流程以0.3mol/L二-(2-乙基己基)磷酸【HDEHP,结构式见以下(1)】的二异丙基苯溶液为有机相,以1.0mol/L乳酸和0.05mol/L二乙基三胺五乙酸【DTPA,结构式见以下(2)】的HNO3溶液为水相。水相含N配体DTPA作为一种掩蔽剂,会优先与锕系配位,生成水溶性配合物保留在水相中,而含O配体HDEHP则将镧系萃入到有机相中,从而实现锕系和镧系的分离。该研究表明,这种流程可使Am3+和Eu3+的分离因子SFAm/Eu达到10以上(Nilsson M et al.,Solvent Extr.Ion Exch.,2007,25:665-701)。虽然TALSPEAK流程有较好的分离效果,但该流程水相络合剂用量较大,且还需使用乳酸缓冲溶液,导致流程变得复杂。此外,由于在高放废液中镧系的含量远大于锕系,更为合理的方法应是将量多的镧系掩蔽在水相中,同时将量少的锕系元素萃至有机相中。因此,该流程采用萃取量大的镧系、掩蔽量小的锕系的方法在实际应用中并不十分合理。
Cyanex 301流程是以工业品Cyanex 301中的主要成分二-(2,4,4-三甲基戊基)二硫代次膦酸【HBTMPDTP,见以下结构式(3)】为萃取剂,以饱和烃类溶剂为稀释剂,在pH 3.5的条件下,对Am3+和镧系进行萃取。研究发现,Cyanex 301对Am3+有很好的选择性,SFAm/Eu值高达5900(Zhu YJ et al.,Solvent Extr.Ion Exch.,1996,14:61-68)。虽然该萃取剂对镧/锕分离有非常好的效果,但存在化学稳定性与辐照稳定性差、产生二次废物量多、不能在较高酸度下萃取等缺点,尚不能用于实际应用中。
BTPs流程主要以2,6-bis(5,6-dialkyl-1,2,4-triazine-3-yl)-pyridine(BTPs)和6,6'-bis(5,6-dialkyl-1,2,4-triazine-3-yl)-2,2'-bypyridine(BTBPs)等含N配体【见以下结构式(4)和结构式(5)】作萃取剂。在单级萃取实验中,SFAm/Eu值可达100左右(Frechette M et al.,Inorg.Chem.,1995,34:3520-3527)。虽然BTPs、BTBPs类萃取剂仅含有C、H、O、N四种元素,理论上可完全焚烧,有利于减少二次污染,但由于其结构中含有氮杂环,化学与辐照稳定性较差,加之萃取动力学缓慢(约1~2h),目前也尚难满足实际应用的需要。
综上所述,虽然目前已有许多用于镧系/锕系分离的流程,但在实际应用中仍存在诸多问题,还不能应用于实际工业环境中,因此开发新型镧系/锕系分离萃取体系显得十分必要。
N,N,N',N',N",N"-六正辛基-氮川三乙酰胺,【N,N,N',N',N",N"-Hexaoctylnitrilotriacetamide,简写为NTAamide(C8),见以下结构式(6)】是一种新型的既不含S、又非N杂环的含N配体,其不仅对Am3+/Eu3+的萃取有一定的选择性(SFAm/Eu=9.67)(Sasaki Y et al.,Chem.Lett.,2013,42:91-92),而且NTAamide(C8)还有如下优点:(1)相比于含S类萃取剂,NTAamide(C8)可有效避免二次废物污染问题;(2)由于NTAamide(C8)萃取剂不含有S元素和N杂环结构,可以从根本上克服化学、辐照稳定性较差等问题;(3)NTAamide(C8)萃取动力学很快,可以在10min内达到萃取平衡。因此NTAamide(C8)是一种非常有应用前景的镧/锕分离萃取剂。不过,较之BTPs、BTBPs等含N杂环类萃取剂,NTAamide(C8)萃取Am3+/Eu3+的分离因子尚不够高【BTPs、BTBPs的SFAm/Eu值可达~100,而NTAamide(C8)的SFAm/Eu值仅~10,其镧锕分离效果在诸多萃取剂中也只处于中等水平】,要直接用于分离难度极大的三价镧/锕元素,显得很不尽如人意,因此还需要对NTAamide(C8)的萃取体系进行改进。
注:结构式中n-C8H17表示正辛基
技术实现要素:
针对目前实际应用中缺少有效分离镧系/锕系的萃取分离体系的技术现状,本发明的首要目的旨在提供一种双二甘酰胺配体。
本发明的次要目的是提供一种制备上述双二甘酰胺配体的方法。
本发明的再一目的是提供含有上述双二甘酰胺配体的镧系/锕系分离萃取体系,该体系不仅可以避免产生二次废物污染,具有较好的化学与辐照稳定性,良好的萃取动力学性能,而且还可以有效提高了镧系/锕系元素的分离萃取效果。
为了达到上述目的,本发明首先提供了一种双二甘酰胺配体,该配体的结构通式如下:
式中R1、R2为H原子、甲基或乙基,R3为H原子或甲基,所述R1、R2相同或不同;X为碳原子数2~4的亚烷基。
本发明提供的制备上述双二甘酰胺配体的方法,该方法首先是在较低温度下通过胺解反应,将结构式(7)的二甘酸酐转化为结构式(8)的单氧杂酰胺羧酸,然后再经二胺类化合物与单氧杂酰胺羧酸间的缩合反应,得到结构式(9)的最终产物双二甘酰胺配体,其合成路线如下:
其步骤和条件如下:
(1)将二甘酸酐加入溶剂Ⅰ中溶解,并置于0~10℃的冰水浴中边搅拌边滴加与二甘酸酐摩尔比为1:(1~1.1)的胺类化合物,滴加完毕后,在搅拌条件下于室温反应1~3h,然后旋蒸除去溶剂I,再向除去溶剂Ⅰ的反应产物中滴加体积浓度为50%的盐酸水溶液,直至晶体析出,继后进行抽滤、重结晶纯化即得白色片状固体的单氧杂酰胺羧酸;
(2)在搅拌条件下,于室温将单氧杂酰胺羧酸、二胺类化合物、缚酸剂按摩尔比4:(1~2):4加入到溶剂Ⅱ中至完全溶解后,再向溶解后的溶液中加入与单氧杂酰胺羧酸等物质的量的缩合剂和催化剂,并在搅拌条件于室温反应至少10h得到含有双二甘酰胺配体的混合液;
(3)将步骤(2)反应所得混合液依次用酸溶液、碱溶液、去离子水洗涤,并分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去溶剂II,然后再采用柱层析纯化即得到淡黄色固体或液体产物,即双二甘酰胺配体。
上述双二甘酰胺配体的制备方法步骤(1)的目的是通过胺解反应得到单氧杂酰胺羧酸。其中所述胺类化合物为二乙胺、甲乙胺、乙胺或二甲胺中的任一种;所述溶剂I为1,4-二氧六环、二氯甲烷、乙醚、四氢呋喃、苯或甲苯中的任一种,优选1,4-二氧六环和四氢呋喃。本步骤中盐酸水溶液的使用量为至少使白色晶体析出完全。本步骤中是采用体积比1:1的甲醇/水的混合溶剂对白色晶体进行重结晶。
上述双二甘酰胺配体的制备方法中步骤(2)的目的是通过缩合反应得到双二甘酰胺配体混合液,其中所述二胺类化合物为乙二胺、丙二胺、丁二胺、N,N'-二甲基乙二胺或N,N'-二甲基-1,4-丁二胺中的任一种;所述溶剂Ⅱ为二氯甲烷、乙酸乙酯、三氯甲烷、四氢呋喃、苯或甲苯中的任一种,优选乙酸乙酯和二氯甲烷;所述缚酸剂可提高反应产率,其为三乙胺、氢氧化钾或碳酸钾中的任一种;所述缩合剂可促进反应进行,其为1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(EDCI)或二环己基碳二亚胺(DCC);所述催化剂可加快反应速率,其为1-羟基苯并三唑(HOBT)或4-二甲氨基吡啶(DMAP)。
上述双二甘酰胺配体的制备方法中步骤(3)的目的是对含有双二甘酰胺配体的混合液进行洗涤,去除其中大部分的杂质、未反应的原料以及副产物,以提高双二甘酰胺配体的纯度。具体为首先向混合液中添加1.0mol/L的酸溶液(例如盐酸溶液、硫酸溶液、硝酸溶液等),静置至含有双二甘酰胺配体的有机相与含有酸的水相分层,分离出有机相,然后向分离出的有机相中添加1.0mol/L碱溶液(例如氢氧化钠溶液、氢氧化钾溶液等),利用碱中和有机相中的酸生成易溶于水的盐,从而除去可能存在有机相中的酸,静置至含有双二甘酰胺配体的有机相与含有碱和/或盐的水相分层,分离出有机相,最后向分离出的有机相中添加去离子水,利用去离子水尽量洗涤出可能存在于有机相中的盐或碱,静置至含有双二甘酰胺配体的有机相与水相分层,分离出有机相。每种溶液至少洗涤一次。
上述双二甘酰胺配体的制备方法中步骤(4)所述干燥是采用无水无机盐进行干燥,以除去步骤(3)所得有机相中的水分,所述无水无机盐为无水硫酸钠、无水硫酸镁或无水氯化钙中的任一种;所述柱层析采用的固定相为300~400目的硅胶,流动相为由甲醇与二氯甲烷并按照体积比1:(10~20)混合而成。
本发明提供的含有上述双二甘酰胺配体的镧系/锕系分离萃取体系,该萃取体系是由有机相和水相等体积混合而成,所述有机相中包含摩尔浓度为0.1~0.7mol/L的N,N,N',N',N",N"-六正辛基-氮川三乙酰胺【NTAamide(C8)】作为萃取剂,优选摩尔浓度为0.4~0.6mol/L;所述水相中包含摩尔浓度为0.005~0.02mol/L的双二甘酰胺配体作为掩蔽剂,优选摩尔浓度为0.01~0.015mol/L,且水相中还含有待分离的镧系和锕系。
上述镧系/锕系分离萃取体系中所述有机相为NTAamide(C8)的长碳链烷烃类化合物溶液,其中长碳链烷烃类化合物是作为稀释剂,对其没有严格要求,其一般为碳原子数为6~12的正链烷烃中的任一种,例如正己烷、正庚烷、正辛烷、正壬烷、正癸烷、正十一烷或正十二烷。
上述镧系/锕系分离萃取体系中所述水相为双二甘酰胺配体的硝酸水溶液,硝酸水溶液中硝酸的摩尔浓度为1.0×10-4~2.0mol/L。
上述镧系/锕系分离萃取体系中有机相和水相于室温搅拌0.25~2h,优选0.5~1h;搅拌结束后采用常规离心手段对有机相和水相进行分离,从而实现镧系元素和锕系元素的分离。
上述镧系/锕系分离萃取体系可以用于实现分离的镧系元素包括镧、铈、镨、钕、钐、铕、钆、铽、镝、钬、铒、铥、镱和镥,锕系元素包括钍、铀、镅和锔。
与现有技术相比,本发明具有以下有益效果:
1、由于本发明制备的双二甘酰胺配体为短碳链水溶性配体,因而与传统的长碳链脂溶性的双二甘酰胺配体相比,更适合溶于水中作为水相掩蔽剂。
2、由于本发明用于萃取体系中的锕系的萃取剂NTAamide(C8)具有独特的非N杂环的三角结构,同时仅含有C、H、O、N四种元素,因而不仅可以极大地提高分离萃取体系的耐辐照稳定性,而且不会产生二次污染物,有利于环境保护。
3、由于本发明提供的萃取体系中是以水溶性双二甘酰胺配体为掩蔽剂,其具有更倾向于与镧系络合的特点,因而可以有效地将镧系掩蔽在水相中,实现锕系和镧系的选择性分离。
4、由于本发明提供的萃取体系对稀释剂的要求并不十分严格,只要非极性的烷基类均可作为稀释剂,因而与传统的镧系/锕系分离萃取体系相比,可以扩大分离萃取体系的适用范围。
5、由于本发明提供的镧系/锕系分离萃取体系可以在工业应用的酸度范围内(0.1~0.5mol/L HNO3)实现良好的分离效果,因而不仅具有较强的实用性,且在先进核燃料循环领域也具有很好的应用前景。
附图说明
图1为本发明实施例1制备的N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)的红外图谱;
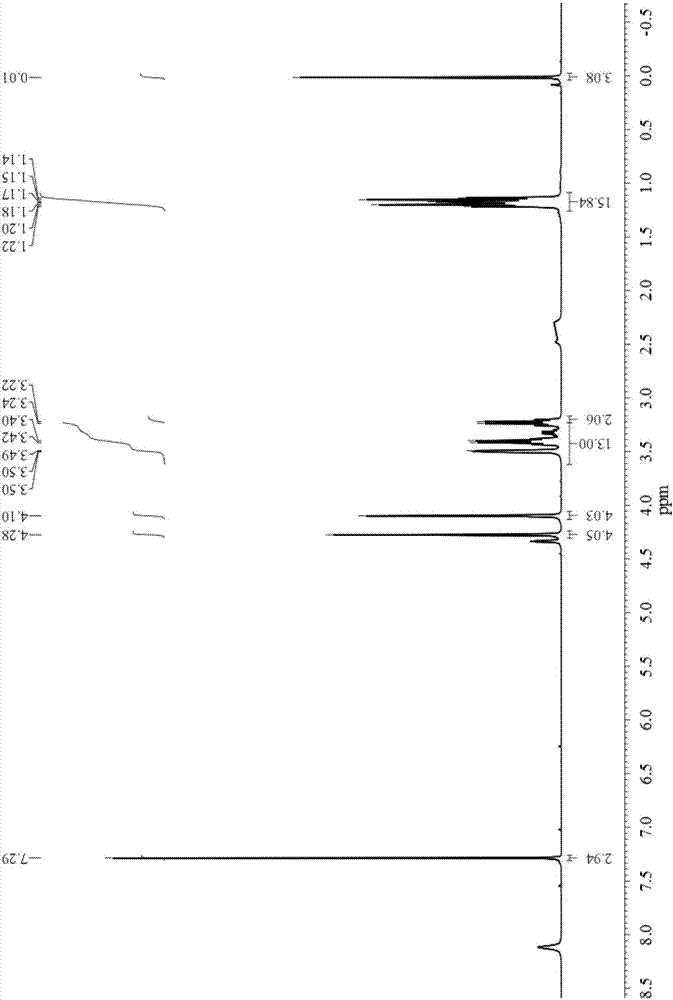
图2为本发明实施例1制备的N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)的1H NMR图谱;
图3为本发明实施例1制备的N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)的13C NMR图谱;
图4为本发明实施例1制备的N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)的质谱图。
具体实施方式
以下将结合附图对本发明各实施例的技术方案进行清楚、完整的描述,显然,所描述实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施例,都属于本发明所保护的范围。
实施例1
本实施例制备的是结构式为(10)的N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA),其合成路线如下所示:
上述TEE-BisDGA的制备过程包括以下步骤:
(1)将二甘酸酐(2.9g,0.025mol)加入40mL四氢呋喃中,并置于0℃的冰水浴中边搅拌边滴加二乙胺(1.8g,0.025mol),滴加完毕后,撤去冰水浴,在搅拌条件下于25℃反应3h;然后旋蒸反应所得产物除去四氢呋喃,再向除去四氢呋喃的反应所得产物中滴加体积浓度为50%的盐酸水溶液,析出白色晶体,对白色晶体进行抽滤、并利用体积比1:1的甲醇/水的混合溶剂进行重结晶纯化得白色片状固体的N,N-二乙基-3-氧戊二酰胺酸;
(2)在搅拌条件下,于25℃将N,N-二乙基-3-氧戊二酰胺酸(4.7g,0.025mol)、乙二胺(0.7g,0.010mol)、三乙胺(2.5g,0.025mol)加入到120mL二氯甲烷中;待各原料完全溶解后,再向溶解后的溶液中加入EDCI(4.8g,0.025mol)和HOBT(3.4g,0.025mol),并在搅拌条件于25℃反应24h得到混合液;
(3)将步骤(2)反应所得混合液依次用1.0mol/L HCl溶液、1.0mol/L NaOH溶液、去离子水洗涤(每种溶液洗涤一次),分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去二氯甲烷,然后对旋蒸所得产物采用柱层析(以300目的硅胶作为固定相,由甲醇和二氯甲烷按照体积比1:20混合而成的溶液作为流动相)纯化得到淡黄色固体产物,即TEE-BisDGA(产率为80%,纯度为99%)。
为了对本实施例制备的N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)的结构进行表征,对其进行了红外测试、核磁共振测试和质谱测试。
本实施例对N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)进行红外测试结果如图1所示,从图中可以看出,其峰所对应的归属分别为胺基基团(-NH-)3309(b,s),亚甲基基团(-CH2-)2978(m),2937(b,m),酰胺基团(-CONH-)1659(s),醚氧基团(-C-O-C-)1271(s),符合目标产物的结构。
本实施例对N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)进行1H NMR测试,其测试条件为仪器频率400MHz,以TMS作为内标,以CDCl3作为溶剂,单位ppm;1H NMR图谱如图2所示,测试结果为δ1.18(12H,m,NCH2CH3),3.23(8H,q,NCH2CH3),3,41(4H,q,NHCH2CH2NH),4.28(8H,s,OCH2CO),8.12(2H,s,NH),符合目标产物的结构。
本实施例对N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)进行13C NMR测试,其测试条件为仪器频率100MHz,以TMS作为内标,以CDCl3作为溶剂,单位ppm;13C NMR图谱如图3所示,测试结果为δ12.9,40.5,41.0,71.7,76.7,167.9,170.1,符合目标产物的结构。
本实施例对N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)进行质谱测试,质谱图如图4所示,测试结果为(质核比m/z)403.2555,[M+H]+,calculated:403.2556(加H+峰);425.2309,[M+Na]+,calculated:425.2376(加Na+峰);441.2013,[M+K]+,calculated:441.2115(加K+峰),符合目标产物的结构。
从上述分析可以看出,N,N,N”',N”'-四乙基-N',N”-乙二基-双二甘酰胺(TEE-BisDGA)的组成与分子结构通过红外、核磁共振1H NMR和13C NMR以及质谱测定得到确认。
实施例2
本实施例制备的是结构式为(11)的N,N,N”',N”'-四乙基-N',N”-丙二基-双二甘酰胺(TEP-BisDGA),其合成路线如下所示:
上述TEP-BisDGA的制备过程包括以下步骤:
(1)将二甘酸酐(2.9g,0.025mol)加入40mL 1,4-二氧六环中,并置于2℃的冰水浴中边搅拌边滴加二乙胺(2.0g,0.027mol),滴加完毕后,撤去冰水浴,在搅拌条件下于25℃反应2.5h;然后旋蒸反应所得产物除去1,4-二氧六环,再向除去1,4-二氧六环的反应所得产物中滴加体积浓度为50%的盐酸水溶液,析出白色晶体,对白色晶体进行抽滤、并利用体积比1:1的甲醇/水的混合溶剂进行重结晶纯化得白色片状固体的N,N-二乙基-3-氧戊二酰胺酸;
(2)在搅拌条件下,于25℃将N,N-二乙基-3-氧戊二酰胺酸(4.7g,0.025mol)、丙二胺(0.5g,0.006mol)、氢氧化钾(1.4g,0.025mol)加入到90mL乙酸乙酯中;待各原料完全溶解后,再向溶解后的溶液中加入EDCI(4.8g,0.025mol)和HOBT(3.4g,0.025mol),并在搅拌条件于25℃反应22h得到混合液;
(3)将步骤(2)反应所得混合液依次用1.0mol/L HNO3溶液、1.0mol/L KOH溶液、去离子水洗涤(每种溶液洗涤一次),分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去乙酸乙酯,然后对旋蒸所得产物采用柱层析(以300目的硅胶作为固定相,由甲醇和二氯甲烷按照体积比1:18混合而成的溶液作为流动相)纯化得到淡黄色固体产物,即TEP-BisDGA(产率为78%,纯度为98%)。
实施例3
本实施例制备的是结构式为(12)的N,N”'-二甲基-N,N”'-二乙基-N',N”-丁二基-双二甘酰胺(DMDEB-BisDGA),其合成路线如下所示:
上述DMDEB-BisDGA的制备过程包括以下步骤:
(1)将二甘酸酐(2.9g,0.025mol)加入30mL二氯甲烷中,并置于4℃的冰水浴中边搅拌边滴加甲乙胺(1.5g,0.025mol),滴加完毕后,撤去冰水浴,在搅拌条件下于25℃反应2.5h;然后旋蒸反应所得产物除去二氯甲烷,再向除去二氯甲烷的反应所得产物中滴加体积浓度为50%的盐酸水溶液,析出白色晶体,对白色晶体进行抽滤、并利用体积比1:1的甲醇/水的混合溶剂进行重结晶纯化得白色片状固体的N-甲基-N-乙基-3-氧戊二酰胺酸;
(2)在搅拌条件下,于25℃将N-甲基-N-乙基-3-氧戊二酰胺酸(4.4g,0.025mol)、丁二胺(0.8g,0.008mol)、碳酸钾(3.5g,0.025mol)加入到160mL苯中;待各原料完全溶解后,再向溶解后的溶液中加入DCC(5.2g,0.025mol)和DMAP(3.0g,0.025mol),并在搅拌条件于25℃反应20h得到混合液;
(3)将步骤(2)反应所得混合液依次用1.0mol/L HCl溶液、1.0mol/L NaOH溶液、去离子水洗涤(每种溶液洗涤一次),分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去苯,然后对旋蒸所得产物采用柱层析(以300目的硅胶作为固定相,由甲醇和二氯甲烷按照体积比1:16混合而成的溶液作为流动相)纯化得到淡黄色固体产物,即DMDEB-BisDGA(产率为78%,纯度为99%)。
实施例4
本实施例制备的是结构式为(13)的N,N”'-二乙基-N',N”-乙二基-双二甘酰胺(DEE-BisDGA),其合成路线如下所示:
上述DEE-BisDGA的制备过程包括以下步骤:
(1)将二甘酸酐(2.9g,0.025mol)加入20mL乙醚中,并置于6℃的冰水浴中边搅拌边滴加乙胺(1.2g,0.027mol),滴加完毕后,撤去冰水浴,在搅拌条件下于25℃反应2.5h;然后旋蒸反应所得产物除去乙醚,再向除去乙醚的反应所得产物中滴加体积浓度为50%的盐酸水溶液,析出白色晶体,对白色晶体进行抽滤、并利用体积比1:1的甲醇/水的混合溶剂进行重结晶纯化得白色片状固体的N-乙基-3-氧戊二酰胺酸;
(2)在搅拌条件下,于25℃将N-乙基-3-氧戊二酰胺酸(4.0g,0.025mol)、乙二胺(0.6g,0.01mol)、三乙胺(2.5g,0.025mol)加入到110mL甲苯中;待各原料完全溶解后,再向溶解后的溶液中加入EDCI(4.8g,0.025mol)和HOBT(3.4g,0.025mol),并在搅拌条件于25℃反应22h得到混合液;
(3)将步骤(2)反应所得混合液依次用1.0mol/L H2SO4溶液、1.0mol/L NaOH溶液、去离子水洗涤(每种溶液洗涤一次),分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去甲苯,然后对旋蒸所得产物采用柱层析(以400目的硅胶作为固定相,由甲醇和二氯甲烷按照体积比1:14混合而成的溶液作为流动相)纯化得到淡黄色固体产物,即DEE-BisDGA(产率为62%,纯度98.5%)。
实施例5
本实施例制备的是结构式为(14)的N,N,N”',N”'-四甲基-N',N”-二甲基-N',N”-乙二基-双二甘酰胺(TMDME-BisDGA),其合成路线如下所示:
上述TMDME-BisDGA的制备过程包括以下步骤:
(1)将二甘酸酐(2.9g,0.025mol)加入20mL苯中,并置于8℃的冰水浴中边搅拌边滴加二甲胺(1.2g,0.0275mol),滴加完毕后,撤去冰水浴,在搅拌条件下于25℃反应1.5h;然后旋蒸反应所得产物除去苯,再向除去苯的反应所得产物中滴加体积浓度为50%的盐酸水溶液,析出白色晶体,对白色晶体进行抽滤、并利用体积比1:1的甲醇/水的混合溶剂进行重结晶纯化得白色片状固体的N,N-二甲基-3-氧戊二酰胺酸;
(2)在搅拌条件下,于25℃将N,N-二甲基-3-氧戊二酰胺酸(4.0g,0.025mol)、N,N'-二甲基乙二胺(0.7g,0.0075mol)、三乙胺(2.5g,0.025mol)加入到75mL三氯甲烷中;待各原料完全溶解后,再向溶解后的溶液中加入EDCI(4.8g,0.025mol)和HOBT(3.4g,0.025mol),并在搅拌条件于25℃反应10h得到混合液;
(3)将步骤(2)反应所得混合液依次用1.0mol/L HCl溶液、1.0mol/L NaOH溶液、去离子水洗涤(每种溶液洗涤一次),分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去三氯甲烷,然后对旋蒸所得产物采用柱层析(以400目的硅胶作为固定相,由甲醇和二氯甲烷按照体积比1:12混合而成的溶液作为流动相)纯化得到淡黄色固体产物,即TMDME-BisDGA(产率为54%,纯度99.5%)。
实施例6
本实施例制备的是结构式为(15)的N,N,N”',N”'-四甲基-N',N”-二甲基-N',N”-丁二基-双二甘酰胺(TMDMB-BisDGA),其合成路线如下所示:
上述TMDMB-BisDGA的制备过程包括以下步骤:
(1)将二甘酸酐(2.9g,0.025mol)加入20mL甲苯中,并置于10℃的冰水浴中边搅拌边滴加二甲胺(1.2g,0.0275mol),滴加完毕后,撤去冰水浴,在搅拌条件下于25℃反应1h;然后旋蒸反应所得产物除去甲苯,再向除去甲苯的反应所得产物中滴加体积浓度为50%的盐酸水溶液,析出白色晶体,对白色晶体进行抽滤、并利用体积比1:1的甲醇/水的混合溶剂进行重结晶纯化得白色片状固体的N,N-二甲基-3-氧戊二酰胺酸;
(2)在搅拌条件下,于25℃将N,N-二甲基-3-氧戊二酰胺酸(4.0g,0.025mol)、N,N'-二甲基-1,4-丁二胺(1.4g,0.0125mol)、三乙胺(2.5g,0.025mol)加入到220mL四氢呋喃中;待各原料完全溶解后,再向溶解后的溶液中加入EDCI(4.8g,0.025mol)和HOBT(3.4g,0.025mol),并在搅拌条件于25℃反应15h得到混合液;
(3)将步骤(2)反应所得混合液依次用1.0mol/L HCl溶液、1.0mol/LNaOH溶液、去离子水洗涤(每种溶液洗涤一次),分离出有机相;
(4)将有机相干燥除去所含水分,并旋蒸除去四氢呋喃,然后对旋蒸所得产物采用柱层析(以400目的硅胶作为固定相,由甲醇和二氯甲烷按照体积比1:10混合而成的溶液作为流动相)纯化得到淡黄色固体产物,即TMDMB-BisDGA(产率为45%,纯度99%)。
应用例7~25
应用例7~25用于举例说明萃取体系的制备以及所制备的萃取体系中镧系/锕系的分离效果。
应用例7~25的萃取体系包括有机相和水相,有机相为NTAamide(C8)的长碳链烷烃类化合物溶液,长碳链烷烃类化合物为碳原子数为6~12的正链烷烃中的任一种;水相为双二甘酰胺配体的硝酸水溶液,双二甘酰胺配体为由实施例1、实施例2、实施例4制备得到,硝酸水溶液中含有241Am和152Eu(其中示踪量的为241Am或152Eu)。应用例7~25中有机相和水相组成见表1所示。
应用对比例1~7的萃取体系与应用例19~25的萃取体系基本相同,其不同在于应用对比例1~7中水相为二酰胺荚醚TEDGA【N,N,N,N-四乙基-3-氧戊酰胺,见结构式(16)】的硝酸水溶液,二酰胺荚醚TEDGA的浓度为0.01mol/L(具体见表1)。
上述应用例7~25以及应用对比例1~7的萃取体系的萃取过程如下:取1.5mL有机相与1.5mL水相混合得到萃取体系,将萃取体系于25℃搅拌0.25~2.0h(具体见表1),随后离心分相,分别取0.5mL有机相和水相,各加入5mL闪烁液,在液体闪烁计数器内测量计数,经测量计算确定的各应用例及应用对比例的萃取体系的SFAm/Eu值见表2所示。
表1 应用例7~25、对比例1~7中有机相和水相组成以及有机相和水相混合搅拌时间
注:为确保测量数据的可靠性以及尽可能降低放射性元素对操作人员的伤害,Am和Eu的使用量控制在0.5h累计液闪计数11000~13000个。
表2 经测量计算确定的各应用例及应用对比例的萃取体系的SFAm/Eu值
从上述获得的各应用例和各应用对比例获得的的萃取体系的SFAm/Eu值可以看出,在最接近于实际工业环境中的酸度(0.1~0.5mol/L)范围内,本发明提供的以NTAamide(C8)为萃取剂(摩尔浓度在0.4~0.6mol/L)、以本发明提供的双二甘酰胺配体为掩蔽剂(摩尔浓度在0.01~0.015mol/L)的萃取体系对Am和Eu的分离效果更好;特别是当酸度越低时,其对Am和Eu的分离效果还具有大幅度提升。然而,当双二甘酰胺配体浓度增大时,其对Am和Eu都具有较强地掩蔽效果,从而降低该体系对Am和Eu的选择性,难以实现对Am和Eu的有效分离,因此双二甘酰胺配体的优选摩尔浓度为0.01~0.015mol/L。
上述是应用例7~25以分离酸性溶液中的Am和Eu为例进行说明,由于锕系中需要处理的最关键元素为Am,而与其物理化学性质最相近的镧系是Eu,如能将Am和Eu较好地分离,则可以很容易实现其它锕系和镧系的分离。
- 还没有人留言评论。精彩留言会获得点赞!