人源肝细胞中Nrf1β基因定向敲除识别序列对、Talens、载体对及应用的制作方法
本发明属于基因敲除技术领域,具体涉及人源肝细胞中nrf1β基因定向敲除识别序列对、talens、载体对及应用。
背景技术:
核因子e2相关因子1(tcf11/nrf1)具有抗氧化、抗炎症、调控蛋白质降解、糖脂代谢平衡等多种重要生理功能。但由于细胞中nrf1蛋白存在多个亚型包括nrf1α、nrf1β、nrf1χ、nrf1d等以及多种翻译后修饰,使得nrf1的功能未能得到透彻的研究。肝细胞系是研究nrf1调控糖脂代谢的良好材料,目前市面上还未见有人源肝细胞系nrf1敲出的细胞系出售,使得nrf1对糖脂代谢调控的具体分子机制和信号通路比较模糊。
传统的研究方法中,使用sirna干扰技术研究nrf1功能的原理是通过降解细胞中nrf1的mrna,从而达到降低细胞中nrf1蛋白质水平,而且不能完全消除细胞中的nrf1,因此得出的研究结果不能排除残留的nrf1蛋白的影响,此外sirna技术还存在脱靶问题,sirna技术的稳定性以及可重复性较差;利用同源重组系统基因敲出技术研究nrf1则是在基因组上将nrf1基因全部敲出,但此方法操作繁琐,在基因组上敲出片段较长可能导致转录失控,并且不能区分不同nrf1亚基的功能。
如何使nrf1的研究变的方便快捷,在不对基因组做出较大长度改变得遗传背景下达到nrf1全部敲出,并且让研究结果稳定可靠可重复,是nrf1研究领域亟待解决的问题。
技术实现要素:
针对现有技术存在的上述不足,本发明要解决的技术问题是:如何提供人源肝细胞中nrf1β基因定向敲除识别序列对、talens、载体对及应用,使该方法操作方便快捷,在不对基因组做出较大长度改变的遗传背景下达到将nrf1β基因全部敲除,使肝细胞中不残留nrf1β蛋白。
为了解决上述技术问题,本发明采用如下技术方案:基于talen的人源肝细胞中nrf1β基因定向敲除识别序列对,所述识别序列对由左臂识别序列和右臂识别序列组成;所述左臂识别序列的核苷酸序列如seqidno.1所示,所述右臂识别序列的核苷酸序列如seqidno.2所示。
用于定向敲除人源肝细胞中nrf1β基因的转录激活样效应因子核酸酶talens,所述转录激活样效应因子核酸酶talens由识别seqidno.1核苷酸序列的核酸酶左臂nrf1β-talenl和识别seqidno.2核苷酸序列的核酸酶右臂nrf1β-talenr组成;所述nrf1β-talenl由识别seqidno.1核苷酸序列的tale-l和非特异性核酸内切酶fokl融合而成,所述nrf1β-talenr由识别seqidno.2核苷酸序列的tale-r和非特异性核酸内切酶fokl融合而成;所述tale-l的氨基酸序列如seqidno.3所示,所述tale-r的氨基酸序列如seqidno.4所示。
用于定向敲除人源肝细胞中nrf1β基因的载体对,所述载体对由nrf1β-talenl载体和nrf1β-talenr载体组成;所述nrf1β-talenl载体包含编码所述tale-l的核苷酸序列,所述nrf1β-talenr载体包含编码所述tale-r的核苷酸序列;所述nrf1β-talenl载体和nrf1β-talenr载体均带有嘌呤霉素抗性基因。
进一步,所述nrf1β-talenl载体为包含编码权利要求2所述tale-l的核苷酸序列的talen-left载体;所述nrf1β-talenr载体为包含编码权利要求2所述tale-r的核苷酸序列的tale-right载体。
所述用于定向敲除人源肝细胞中nrf1β基因的载体对在定向敲除人源肝细胞系中nrf1β基因、构建nrf1β基因敲除型人源肝细胞系中的应用。
进一步,所述人源肝细胞为人源肝细胞hl7702细胞。
nrf1β基因敲除型人源肝细胞系的构建方法,包括如下步骤:
1)采用转染法将所述载体对转染入人源肝细胞中;
2)用嘌呤霉素筛选步骤1)转染培养后的人源肝细胞,得到成功转染nrf1β-talenl载体质粒和nrf1β-talenr载体质粒的人源肝细胞;
3)将步骤2)筛选成功的转染nrf1β-talenl载体质粒和nrf1β-talenr载体质粒的人源肝细胞用胰酶消化悬浮后,继续培养至单细胞克隆长成,挑取由单个细胞长成且细胞状态良好的群落继续扩大培养,得到所述nrf1β基因敲除型人源肝细胞系。
进一步,采用转染法将所述载体对转染入人源肝细胞中的具体步骤为:
a、向细胞培养孔板中每孔加入人源肝细胞,采用dmem培养基对所述人源肝细胞进行培养至所述人源肝细胞在孔板底部生长覆满80%后,将每孔中的dmem培养基吸出并加入opti-mem,将细胞培养孔板中的各个孔分为转染组和对照组备用;
b、取nrf1β-talenl载体质粒、nrf1β-talenr载体质粒和opti-mem混合均匀,得到试剂1;其中,所述nrf1β-talenl载体质粒、nrf1β-talenr载体质粒和opti-mem的质量体积比为1.5μg:3μg:100μl;将opti-mem作为试剂2;将lipo2000和opti-mem以9:100的体积比混合均匀,得到试剂3;
c、将试剂1和试剂3等体积混合并静置15min后,加入步骤a转染组中,培养8h;将试剂2和试剂3等体积混合并静置15min后,加入步骤a对照组中,培养8h;
d、将培养结束后的转染组和对照组中液体吸出,并分别向转染组和对照组孔中各加入含10wt.%fbs的dmem培养基,完成转染步骤。
进一步,步骤2)中所述用嘌呤霉素筛选步骤1)转染培养后的人源肝细胞的具体步骤为:将嘌呤霉素加入含10wt.%fbs的dmem培养基中混合均匀,得到含嘌呤霉素培养基,其中,所述嘌呤霉素在所述含嘌呤霉素培养基中的终浓度为2μg/ml;在人源肝细胞转染nrf1β-talenl载体质粒和nrf1β-talenr载体质粒48h后,用所述含嘌呤霉素培养基继续培养至对照组中人源肝细胞全部死亡为止,a孔中仍剩余存活的人源肝细胞即为成功转染nrf1β-talenl载体质粒和nrf1β-talenr载体质粒的人源肝细胞。
进一步,所述人源肝细胞为人源肝细胞hl7702细胞。
相比现有技术,本发明具有如下有益效果:
1、本发明利用talen技术,使基因编辑变的简单易行,可对任意核酸碱基位点进行编辑,通过敲出非3倍数的碱基形成译码突变来实现基因编辑,在基因组上只有数个碱基的插入或缺失,却能在蛋白水平上完全消除目的基因的表达。具体地,本发明利用talen技术在人源肝细胞hl7702细胞中编辑nrf1基因组碱基序列,通过单克隆细胞系的筛选,培养和鉴定成功分别制造出nrf1β敲除的稳定单克隆细胞系7b2。单克隆细胞系在基因组上将nrf1的cds区域缺失若干碱基,使nrf1蛋白因编码区碱基的移码缺失而在蛋白水平上完全敲出,实现了对基因组极小的改变达到nrf1β蛋白的完全敲出,取得了优良的基因编辑效果。
2、本发明基因敲除的7b2细胞系是通过单克隆培养挑选技术获得的单细胞克隆细胞系,经过长达数月的培养,发现得到的7b2细胞系状态优良形态稳定,说明本发明得到的基因敲除细胞系稳定性良好。
3、本发明利用7b2细胞系研究肝细胞中nrf1β功能,只需将7b2细胞系与对照细胞系共同对照处理即可,使得转录因子nrf1功能研究简单易行,且能节省大量昂贵的生化试剂,本发明7b2是nrf1β蛋白完全敲出的细胞系,得出的研究结果能完全体现出nrf1β在肝细胞中的功能,可作为nrf1β在肝细胞中功能研究的绝佳材料,7b2细胞系为nrf1蛋白贝塔亚型敲出的细胞系,可与nrf1蛋白其他亚型敲出的细胞系共同使用以实现对nrf1蛋白各亚型功能的研究。
4、采用本发明方法构建得到的7b2细胞系是单克隆细胞系,遗传表达稳定,得出的研究结果可重复性好,研究结果准确可靠。
附图说明
图1为nrf1β基因编辑位点选定及talen识别位点选定图;
图2为7b2中nrf1基因组nrf1-7b2-1、nrf1-7b2-2与正常nrf1基因组nrf1-wt比对图;
图3为以7b2基因组为模板,pcr扩增出敲出位点序列测序结果图;
图4为定量pcr检测7b2与hl7702细胞中nrf1的mrna水平变化图;
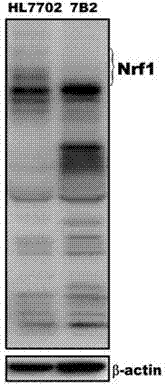
图5为用蛋白质免疫印迹方法检测7b2与hl7702细胞中nrf1蛋白质图。
具体实施方式
下面结合具体实施例对本发明作进一步详细说明。本实施案例在以本发明技术为前提下进行实施,现给出详细的实施方式和具体的操作过程来说明本发明具有创造性,但本发明的保护范围不限于以下的实施例。
实施例1编辑位点的选定及nrf1β-talen质粒的设计和构建
根据talen技术基因编辑原理,选取nrf1基因组中cds区域nrf1β翻译起始密码子处做为nrf1β基因talen靶向编辑位点,序列为:5’-tagtgaacagtggtgctcttctcccctcccaggcc
根据设计的talen左臂和右臂识别的核酸碱基,按照a对应ni,t对应ng,g对应nn,c对应hd的规则,得到识别seqidno.1核苷酸序列的tale-l,所述tale的氨基酸序列如seqidno.3所示(ltpdqvvaias
设计nrf1β敲出位点pcr反应扩增引物序列:
nrf1βf:5-gaagct
nrf1βr:5-ggttggt
nrf1βf引物插入kpni限制性核酸内切酶酶切位点,nrf1βr引物插入bglii限制性核酸内切酶酶切位点。(粗体标注碱基为对应限制性核酸内切酶识别位点。)
按照talen质粒试剂盒使用说明书,将nrf1β-talenl和nrf1β-talenr的核苷酸序列分别插入talen-left(含有嘌呤霉素抗性基因)和tale-right表达载体中,并提取质粒,分别得到nrf1β-talenl载体质粒和nrf1β-talenr载体质粒。
实施例2nrf1β基因敲出的单克隆细胞系的筛选、培养和鉴定
1)nrf1β-talen质粒转染hl7702细胞:
a、将hl7702细胞种六孔板中2个孔,分别记为a(质粒转染组)、b孔(对照组),每孔30万细胞,采用dmem培养基进行培养过夜。待细胞在孔板底部生长至铺满底板80%左右开始准备转染nrf1β-talen质粒。
b、将a、b孔中dmem培养基吸出,加入800μlopti-mem。
c、准备液1、液2和液3;其中,液1配方如下:取nrf1β-talenl质粒1.5μg、nrf1β-talenr质粒3μg加入100μlopti-mem,混匀放置5分钟;
液2:100μlopti-mem;
液3:取lipo200018μl加入200μlopti-mem,混匀放置5分钟;
d、取100μl液3加入液1中,混匀后放置15分钟,加入a孔;
e、取100μl液3加入液2中,混匀后放置15分钟,加入b孔;
f、将步骤d、e加入液体后的孔板放入培养箱中,于体积浓度5%co2、37℃下培养8小时后,吸出各孔中培养基,向a、b孔中各加入2ml含10wt.%fbs的dmem培养基。
2)嘌呤霉素筛选获得成功转染细胞:
配制含嘌呤霉素培养基:将嘌呤霉素加入含10wt.%fbs的dmem培养基中混合均匀,使嘌呤霉素终浓度为2μg/ml。
待步骤1)hl7702细胞转染nrf1β-talen质粒48小时后,用含嘌呤霉素培养基继续培养a、b孔中的细胞,于体积浓度5%co2、37℃下培养3-5天,直到对照组b孔中细胞全部死亡为止,此时a孔中剩余的细胞即为成功转染了nrf1β-talen质粒的hl7702细胞。
3)挑选单克隆细胞系:
将2)中得到的成功转染nrf1β-talen质粒的hl7702细胞用胰酶消化悬浮,具体操作为用无血清dmem培养基清洗培养板中的细胞2次,除去原培养基中的血清残留,吸干培养基,加入2ml胰酶,于37℃培养箱中消化3至5分钟。
待消化结束后,用血球计数板对消化后的细胞计数,根据细胞浓度,吸取100个细胞加入10ml含10wt.%fbs的dmem培养基中并混匀。
将上述混匀后的含细胞培养基种96孔板,每孔接种量100μl,置于培养箱中于体积浓度5%co2、37℃下培养至单细胞克隆长成。
显微镜下观察96孔板中细胞群落,选取由单个细胞长成且细胞状态良好的群落继续扩大培养,得到单克隆细胞系。
4)单克隆细胞系鉴定。
将步骤3)扩大培养的单克隆细胞系及对照细胞系(hl7702)提取基因组、总rna、总蛋白。
用单克隆细胞系基因组做pcr,使用引物nrf1βf和nrf1βr,扩增出nrf1β-talen靶序列片段并测序,检测nrf1基因的碱基编辑效果。
将单克隆细胞系的总rna反转录为cdna,用定量pcr技术检测单克隆细胞系中nrf1的mrna水平变化。
通过蛋白质免疫印迹技术使用nrf1抗体检测单克隆细胞系总蛋白中nrf1蛋白水平变化。
通过上述3种检测方法,鉴定出hl7702细胞中nrf1β成功敲出的单克隆细胞系7b2细胞系,鉴定结果显示,7b2细胞系中nrf1基因缺失5/12个dna碱基(一条染色体上缺失5个碱基,另一条染色体上缺失12个碱基,全都破坏了起始密码子)(图2、图3),7b2细胞系中nrf1在mrna水平的表达上没有显著变化(图4),7b2细胞系中nrf1β蛋白条带完全消失(图5)。结果表明成功获得人源肝细胞hl7702中nrf1β蛋白完全敲出的单克隆细胞系7b2。由上述实验分析可以看出,本实施例成功获得人源肝细胞系中nrf1β蛋白完全敲出单克隆细胞系7b2。
对获得的7b2细胞系进行传代培养时,使用含双抗和10%fbs的高糖dmem培养基培养,培养环境为5%co2,37℃。每2-3天更换新鲜培养基,细胞铺满培养瓶底即可传代。细胞系传代时先用pbs缓冲液清洗贴壁细胞,再使用胰酶消化5-10分钟,加入含10%fbs的高糖dmem培养基终止消化,用吸管将细胞吹散开,一瓶传2瓶或3瓶。
本发明的上述实施例仅仅是为说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其他不同形式的变化和变动。这里无法对所有的实施方式予以穷举。凡是属于本发明的技术方案所引申出的显而易见的变化或变动仍处于本发明的保护范围之列。
sequencelisting
<110>重庆大学;
<120>人源肝细胞中nrf1β基因定向敲除识别序列对、talens、载体对及应用;
<160>6
<170>patentinversion3.5
<210>1
<211>17
<212>dna
<213>人工序列
<220>
<223>seqidno.1的核苷酸序列
<400>1
tggtgctcttctcccct17
<210>2
<211>18
<212>dna
<213>人工序列
<220>
<223>seqidno.2的核苷酸序列
<400>2
tttcacttgctgatgtgt18
<210>3
<211>578
<212>prt
<213>人工序列
<220>
<223>seqidno.3的氨基酸序列
<400>3
ltpdqvvaiasngggkqaletvqrllpvlcqdhgltpdqvvaiasnnggkqaletvqrll60
pvlcqdhgltpdqvvaiasnnggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqa120
letvqrllpvlcqdhgltpdqvvaiasnnggkqaletvqrllpvlcqdhgltpdqvvaia180
shdggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrllpvlcqdhglt240
pdqvvaiashdggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrllpv300
lcqdhgltpdqvvaiasngggkqaletvqrllpvlcqdhgltpdqvvaiashdggkqale360
tvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrllpvlcqdhgltpdqvvaiash420
dggkqaletvqrllpvlcqdhgltpdqvvaiashdggkqaletvqrllpvlcqdhgltpd480
qvvaiashdggkqaletvqrllpvlcqdhgltpdqvvaiashdggkqaletvqrllpvlc540
qdhgltpdqvvaiasngggkqaletvqrllpvlcqdhg578
<210>4
<211>612
<212>prt
<213>人工序列
<220>
<223>seqidno.4的氨基酸序列
<400>4
ltpdqvvaiasngggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrll60
pvlcqdhgltpdqvvaiasngggkqaletvqrllpvlcqdhgltpdqvvaiashdggkqa120
letvqrllpvlcqdhgltpdqvvaiasniggkqaletvqrllpvlcqdhgltpdqvvaia180
shdggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrllpvlcqdhglt240
pdqvvaiasngggkqaletvqrllpvlcqdhgltpdqvvaiasnnggkqaletvqrllpv300
lcqdhgltpdqvvaiashdggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqale360
tvqrllpvlcqdhgltpdqvvaiasnnggkqaletvqrllpvlcqdhgltpdqvvaiasn420
iggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrllpvlcqdhgltpd480
qvvaiasnnggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletvqrllpvlc540
qdhgltpdqvvaiasnnggkqaletvqrllpvlcqdhgltpdqvvaiasngggkqaletv600
qrllpvlcqdhg612
<210>5
<211>27
<212>dna
<213>人工序列
<220>
<223>nrf1βf的核苷酸序列
<400>5
gaagctggtaccggagctgacactgtg27
<210>6
<211>28
<212>dna
<213>人工序列
<220>
<223>nrf1βr的核苷酸序列
<400>6
ggttggtagatctgaaggtggagttgag28
- 还没有人留言评论。精彩留言会获得点赞!
- 用CRISPR/Cas9技术易于筛选获得目的基因敲除细胞系的方法及产品与制造工艺
- 一种Candida amazonensis的FLP/FRT基因敲除方法与制造工艺
- 玉米低镁条件下特异性转运镁离子基因ZmMGT10及其应用的制造方法与工艺
- CRISPR‑Cas9特异性敲除猪SLA‑3基因的方法及用于特异性靶向SLA‑3基因的sgRNA与制造工艺
- 利用CRISPR/Cas9敲除人PD‑1基因构建靶向CD19CAR‑T细胞的方法与制造工艺
- 一种构建基因替代小鼠研究野生型和突变体非肌性肌球蛋白重链II功能的方法与制造工艺
- 构建在海马体区域特异性敲除IKKα基因的小鼠模型的方法及打靶载体和试剂盒与制造工艺
- 猪ApoE基因敲除打靶载体及其构建方法和应用与制造工艺
- 一种血小板减少症的斑马鱼模型的制造方法与工艺
- 基于CRISPR技术敲除FUT8基因的方法与制造工艺