一种甲苯磺酸索拉非尼晶型化合物及其制备方法与流程
本发明属于医药
技术领域:
,涉及一种甲苯磺酸索拉非尼晶型化合物及其制备方法。
背景技术:
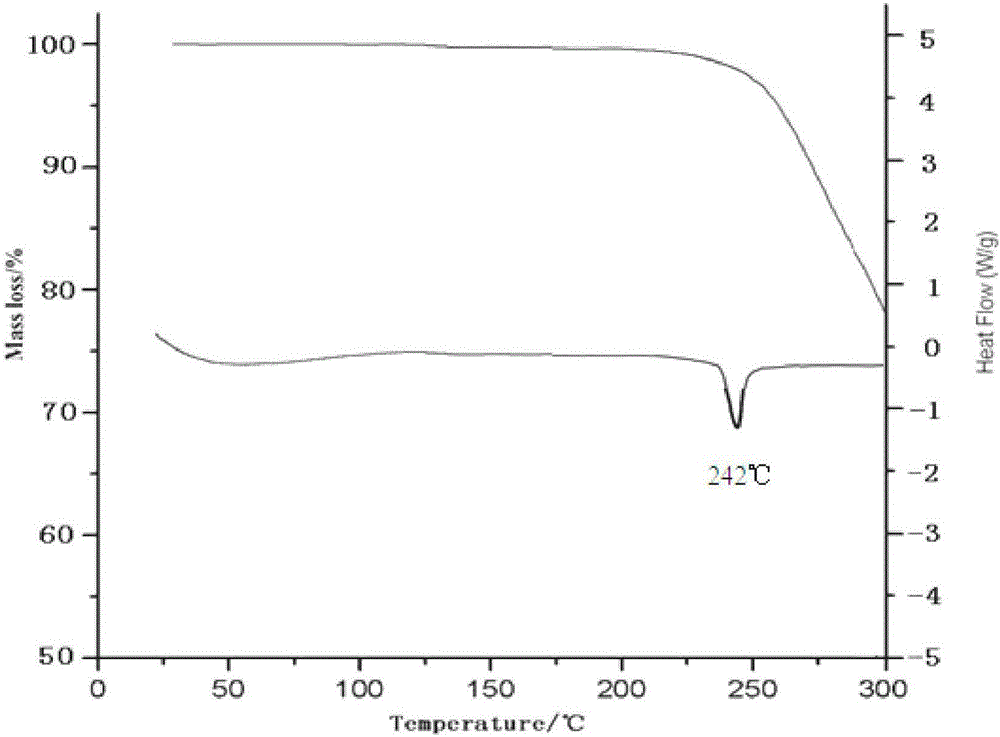
:对甲苯磺酸索拉非尼(sorafenib),化学名称为:N-[4-氯-3-(三氟甲基)苯基]-N′-[4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]脲对甲苯磺酸盐,具有式1所示的化学结构,是由德国拜耳公司和Onxy公司共同研制开发的新型信号转导抑制剂和多靶点抗肿瘤药物。索拉非尼具有双重的抗肿瘤作用:既可通过阻断由RAF/MEK/ERK介导的细胞信号传导通路而直接抑制肿瘤细胞的增殖,还可通过作用于VEGFR,抑制新生血管的形成和切断肿瘤细胞的营养供应而达到遏制肿瘤生长的目的。2006年在中国上市,2008年中国批准其用于晚期肝癌的治疗。2005年12月索拉非尼以其甲苯磺酸盐的形式被美国FDA批准上市,用于先前使用α-干扰素或IL-2没有应答或者不适于这些疗法的晚期肾细胞癌(RCC)患者,商品名为Nexavar;2006年被批准进入中国市场;2006年7月,索拉非尼获得欧盟的上市批准;2007年被欧盟批准用于肝细胞癌的治疗。甲苯磺酸索拉非尼是无味、介于白色和棕色之间的固体。热稳定性良好,不吸水。甲苯磺酸索拉非尼在N,N-二甲基甲酰胺中易溶解,在甲醇中微溶,在乙腈、无水乙醇中极微溶解,在水、二氯甲烷中几乎不溶,溶于聚乙二醇400。拜尔公司公开了甲苯磺酸索拉非尼化合物专利(CN00802685.8),然后又公开了甲苯磺酸索拉非尼多晶型物I、多晶型物II、多晶型物III、甲醇溶剂化物和乙醇溶剂化物等5种晶型(CN200580040775.0),限定了多晶型物I的X射线衍射2θ的值(55个),同时也公开了由多晶型物II制备多晶型物I、多晶型物III、甲醇溶剂化物和乙醇溶剂化物的方法。随后浙江华海药业股份有限公司公开了多晶型A(CN201210190995.3)和索拉非尼游离碱多晶型I(201210249228.5),齐鲁制药有限公司公开了索拉非尼晶型A(CN201210349476.7),上海创诺制药有限公司公开了索拉非尼对甲苯磺酸盐N-甲基吡咯烷酮溶剂化物的多晶型物(NMP-1,NMP-2,NMP-3)。上海北卡医药技术有限公司对甲苯磺酸索拉非尼一水合物的多晶型(CN201511018488.1)。一个原料药的不同多晶型可以有不同的化学和物理特性,包括熔点、化学反应性、表观溶解度、溶解速率、光学和机械性质、蒸气压和密度。这些特性可以直接影响原料药和制剂的处理和/或生产,并且会影响制剂的稳定性、溶解度和生物利用度。当化合物存在多晶型时,由于特定多晶型物具有特异性的热力学性质和稳定性,因此在制备的过程中,了解在各个剂型中应用的化合物的晶型是重要的,以保证生产过程应用相同形态的药物活性化合物。因此,保持药物活性化合物是单一的晶型或是一些晶型的已知混合物是必要的。甲苯磺酸索拉非尼在结晶时,如果采用不同的溶剂和工艺条件,则其分子在各晶型晶胞的排列数目和位置及点阵形式不一样,形成不同的晶体结构,甲苯磺酸索拉非尼多晶型的变化会改变其性质,质量和药效。因此,制备甲苯磺酸索拉非尼的稳定结晶,对于进一步研究该化合物的物理化学性质,研究其药物组合及临床应用,具有十分重要的意义。采用本发明的方法制备的甲苯磺酸索拉非尼无水晶型在晶型转化稳定性,物理稳定性和化学稳定性方面有助于提高其生物利用度,降低不良反应,增加临床疗效。技术实现要素:本发明的首要发明目的在于提出了一种甲苯磺酸索拉非尼晶型化合物及其制备方法。为了实现本发明的目的,采用的技术方案为:本发明提供一种甲苯磺酸索拉非尼晶型化合物,该化合物以2θ±0.2°衍射角表示的X-射线粉末衍射图谱在3.12°、3.81°、4.78°、5.54°、6.82°、7.57°、9.72°、10.63°、11.81°、12.90°、14.23°、16.13°、16.90°、17.52°、18.34°、19.24°、21.15°、22.21°、23.81°、24.76°、25.86°、28.42°、28.75°、29.12°、30.63°和34.35°处显示有特征衍射峰。本发明提供的甲苯磺酸索拉非尼晶型化合物使用Cu-Kα射线测量得到的X-射线粉末衍射图如图1所示。本发明还提供了一种甲苯磺酸索拉非尼晶型化合物的制备方法,具体步骤为:a)将甲苯磺酸索拉非尼粗品溶于混合溶剂A,溶液搅拌加热后使甲苯磺酸索拉非尼粗品全部溶解,至溶液澄清,趁热过滤;b)将上述得到的溶液降温,当降至20~30℃时向溶液中按1.0~2.0mL/min的流速加入预冷的混合溶剂B至出晶,析出晶体,继续降温至-10℃~-5℃,保温搅拌养晶至析晶完全;c)抽滤,收集晶体,少量乙醇洗涤,真空干燥,得到甲苯磺酸索拉非尼结晶。优选地,步骤a)中,所述混合溶剂A为二甲基亚砜和乙醇的混合溶剂,二甲基亚砜和乙醇的体积比为2~3:1;甲苯磺酸索拉非尼和混合溶剂A的质量体积比为1:10~20。优选地,步骤b)中,所述的混合溶剂B为乙醇和石油醚的混合溶剂,乙醇和石油醚的体积比为1:3~5;混合溶剂A与混合溶剂B的体积比为1:2~5。更优选地,步骤b)中,降温幅度为每10分钟1℃~5℃,养晶温度为-10℃~-5℃,养晶时间为0.5~3h。本发明还提供了一种含有本发明甲苯磺酸索拉非尼晶型化合物的药物组合物,该药物组合物为含有甲苯磺酸索拉非尼晶体的片剂。研究表明,在X-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。本发明所提供的甲苯磺酸索拉非尼结晶其粉末X-射线衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型。下面通过对本发明提供的甲苯磺酸索拉非尼晶型化合物进行研究来解释和说明本发明技术方案:1、晶型检测取本发明制备得到的甲苯磺酸索拉非尼结晶,使用Cu-Kα射线测量得到的X-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的X-射线粉末衍射图在3.12°、3.81°、4.78°、5.54°、6.82°、7.57°、9.72°、10.63°、11.81°、12.90°、14.23°、16.13°、16.90°、17.52°、18.34°、19.24°、21.15°、22.21°、23.81°、24.76°、25.86°、28.42°、28.75°、29.12°、30.63°和34.35°处显示有特征峰。2、差热分析及热重分析对本发明制备的甲苯磺酸索拉非尼晶体进行差热和热重分析,结果如附图2所示;结果表明,本品在150℃前没有吸收峰,说明样品中无结晶水或结晶溶剂;本品在242℃处有吸热峰。本品经熔点测定:241~243℃,从侧面证明了其为一种不同的晶型。3、水分分析采用卡式水分测定仪测定,本发明的甲苯磺酸索拉非尼结晶的含水量为0.05%。4、纯度检测经HPLC纯度检测,本发明制备得到的甲苯磺酸索拉非尼结晶的纯度可达到99.7~99.9%。与现有技术相比,本发明具有如下优点:(1)本发明所提供的甲苯磺酸索拉非尼晶型化合物是一种不同于现有技术的新晶型;(2)本发明所提供的甲苯磺酸索拉非尼晶型化合物引湿性得到了改善,稳定性、流动性好,很好地改善了甲苯磺酸索拉非尼在水中的溶解度,提高了生物利用度,有助于药物给药途径的选择设计和药物制剂工艺参数的确定,从而提高药品生产质量;(3)本发明所提供的甲苯磺酸索拉非尼晶型化合物制备方法简单易操作,反应条件温和,适合大规模生产。附图说明图1为本发明实施例1制备的甲苯磺酸索拉非尼晶型化合物的X-射线粉末衍射图谱。图2本发明实施例1制备的甲苯磺酸索拉非尼晶型化合物的TG-DSC图谱。图3是采用本发明甲苯磺酸索拉非尼晶型的片剂的自制制剂和进口制剂在水-1%SDS、pH1.0HCl-1%SDS、pH4.0ABS-1%SDS、pH6.8PBS-1%SDS四种溶出介质中的溶出曲线。具体实施例以下用实施例对本发明的技术方案进行详细说明,将有助于对本发明的技术方案的优点、效果有更进一步的了解,实施例不限定本发明的保护范围,本发明的保护范围由权利要求来决定。实施例1:甲苯磺酸索拉非尼晶型化合物的制备取甲苯磺酸索拉非尼100g于反应瓶中,加入1000ml二甲基亚砜和乙醇的混合溶液(二甲基亚砜和乙醇的体积比为3:1),加热至50℃,搅拌溶清,趁热过滤;边搅拌,边降温至20℃(降温幅度为每10分钟5℃),向溶液中按1.0mL/min的流速加入预冷的混合溶剂B(乙醇和石油醚的体积比为1:3)2000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),搅拌3h。真空抽滤,滤饼于50℃真空干燥6h,得90.3g类白色固体,收率90.3%。实施例2:甲苯磺酸索拉非尼晶型化合物的制备取甲苯磺酸索拉非尼100g于反应瓶中,加入1500ml二甲基亚砜和乙醇的混合溶液(二甲基亚砜和乙醇的体积比为2:1),加热至60℃,搅拌溶清,趁热过滤;边搅拌,边降温至25℃(降温幅度为每10分钟5℃),向溶液中按1.5mL/min的流速加入预冷的混合溶剂B(乙醇和石油醚的体积比为1:5)3000ml至出晶,继续降温至-10℃(降温幅度为每10分钟1℃),搅拌2h。真空抽滤,滤饼于50℃真空干燥4h,得88.6g类白色固体,收率88.6%。所制得晶体的使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。实施例3:甲苯磺酸索拉非尼晶型化合物的制备取甲苯磺酸索拉非尼150g于反应瓶中,加入1500ml二甲基亚砜和乙醇的混合溶液(二甲基亚砜和乙醇的体积比为2:1),加热至45℃,搅拌溶清,趁热过滤;边搅拌,边降温至30℃(降温幅度为每10分钟5℃),向溶液中按2mL/min的流速加入预冷的混合溶剂B(乙醇和石油醚的体积比为1:5)3000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),搅拌2h。真空抽滤,滤饼于50℃真空干燥5h,得137.3g类白色固体,收率91.5%。所制得的甲苯磺酸索拉非尼晶体使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。实施例4:甲苯磺酸索拉非尼晶型化合物的制备取甲苯磺酸索拉非尼80g于反应瓶中,加入800ml二甲基亚砜和乙醇的混合溶液(二甲基亚砜和乙醇的体积比为3:1),加热至45℃,搅拌溶清,趁热过滤;边搅拌,边降温至30℃(降温幅度为每10分钟5℃),向溶液中按2mL/min的流速加入预冷的混合溶剂B(乙醇和石油醚的体积比为1:4)4000ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),搅拌2h。真空抽滤,滤饼于50℃真空干燥5h,得74g类白色固体,收率92.4%。所制得的甲苯磺酸索拉非尼晶体使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。实施例5:甲苯磺酸索拉非尼晶型化合物的制备取甲苯磺酸索拉非尼80g于反应瓶中,加入1600ml二甲基亚砜和乙醇的混合溶液(二甲基亚砜和乙醇的体积比为2:1),加热至45℃,搅拌溶清,趁热过滤;边搅拌,边降温至30℃(降温幅度为每10分钟5℃),向溶液中按2mL/min的流速加入预冷的混合溶剂B(乙醇和石油醚的体积比为1:5)3200ml至出晶,继续降温至-5℃(降温幅度为每10分钟1℃),搅拌2h。真空抽滤,滤饼于50℃真空干燥5h,得72.6g类白色固体,收率90.8%。所制得的甲苯磺酸索拉非尼晶体使用Cu-Kα射线测量得到的X-射线粉末衍射谱图与实施例1相似。下面通过实验例进一步说明本发明:实验例1:流动性实验本实验例采用固定漏斗法测定各实施例样品的休止角,从而评价本发明提供的甲苯磺酸索拉非尼结晶的流动性。具体方法如下:将漏斗置于坐标纸上的适宜高度,取实施例1-5批制备的样品,从固定的漏斗中自由留下,直到形成的圆锥体顶部与漏斗口接触,测算物料堆积层的斜边与水平线的夹角度数(休止角θ)。实验结果如表1所示。表1:流动性实验结果样品12345平均值θ(°)35.734.834.635.635.535.2从表1的实验结果分析,本发明实施例1-5制备得到的甲苯磺酸索拉非尼结晶的流动性很好,有利于提高分装的准确性,并且与其他成分混合时易于混合均匀。实验例2:溶解度测定试验品:本发明实施例1-3所制备的样品;对照品1是市售甲苯磺酸索拉非尼原料药;对照品2是参照专利CN201210349476.7实施例2制备的甲苯磺酸索拉非尼晶型;对照品3是参照专利CN201410003888.4实施例2制备的甲苯磺酸索拉非尼晶型;对照品4是参照专利CN201210190995.3实施例一制备的甲苯磺酸索拉非尼多晶型;对照品5是参照专利CN201210249228.5实施例一制备的甲苯磺酸索拉非尼多晶型;对照品6是参照专利CN201210581826.2实施例3制备的甲苯磺酸索拉非尼多晶型;对照品7是参照专利CN201511018488.1实施例1制备的甲苯磺酸索拉非尼一水物;对照品8是参照专利CN201410717357.1实施例1制备的甲苯磺酸索拉非尼晶型;对照品9是参照专利CN201410390897.3实施例1制备的甲苯磺酸索拉非尼多晶型。参照中国药典2015年版二部凡例测定其溶解性,方法:取本品适量,分别加入水,每隔5分钟强力振摇30秒钟,观察30分钟内的溶解情况,即得,结果见表2。表2本发明的晶型和对照品在水中溶解性试验结果将上述实施例1-3溶解的水溶液样品在25℃恒温搅拌72小时,取样5ml。样品经0.45μm微孔滤膜过滤,弃去初滤液,取续滤液20μL测定药物含量即为水中溶解度(mg/ml)。结果见表3:表3本发明晶型与现有技术晶型在水中的溶解度对比从表2-3可以看出,25℃下,本发明甲苯磺酸索拉非尼晶体化合物的在水中的溶解度与现有技术相比,有显著提高。实验例3:有关物质检测本实验例对实施例1~5所制备的甲苯磺酸索拉非尼结晶中的有关物质杂质进行检测分析,按照中国药典2015版第二部附录ⅪⅩF药品杂质分析指导原则进行,结果见表4。表4:各实施例样品杂质检测分析结果样品总杂(%)最大单杂(%)含量(%)实施例10.470.2499.91实施例20.450.2399.90实施例30.460.2599.88实施例40.480.2499.89实施例50.470.2599.85实验例4、稳定性试验本实验例通过加速试验和长期试验,考察本发明提供的甲苯磺酸索拉非尼结晶的稳定性。1、加速试验取实施例1-3制备的样品,于温度40±2℃、相对湿度75±5%的条件下放置6个月,分别于0、1、2、3、6个月末取样测定性状、有关物质、含量,结果见表5。表5:加速试验结果(温度40±2℃,相对湿度75±5%)从表5看出,本发明甲苯磺酸索拉非尼结晶在温度40±2℃、相对湿度75±5%的条件下放置6个月,有关物质含量没有明显升高,各指标均无明显变化,说明本品稳定性好。2、长期试验取实施例1-3制备的样品,于温度25±2℃、相对湿度60±5%的条件下放置6个月,分别于0、3、6、9、12、18、24个月末取样测定性状、有关物质、含量,结果见表6。表6:长期试验结果(温度25±2℃,相对湿度60±5%)从表6看出,本发明甲苯磺酸索拉非尼结晶在温度25±2℃、相对湿度60±5%的条件下放置24个月稳定,各指标均无明显变化。实施例8:溶出度一致性评价甲苯磺酸索拉非尼是多晶型药物,不同企业原料由于晶型不同,其在水中溶解特性也有差异,为避免由于同一药物不同晶型的特性导致溶出速率、溶解度存在差异,而影响生物利用度或治疗的有效性,通常对制剂进行体内外溶出度一致性评价,以确保药品使用的安全性和有效性。将本发明的甲苯磺酸索拉非尼晶型用普通的万能粉碎机粉碎,过120目筛,按专利200680007187.1的处方制备片剂(规格:200mg,原研制剂的原料采用微粉法制备),按照文献的方法(吴晓刚,等.甲苯磺酸索拉非尼片的制备及体外溶出行为考察.药学及临床研究.2015,23(2):144-146.),分别以水溶液(含1%SDS)、0.1mol·L-1盐酸溶液(含1%SDS)、pH4.0醋酸盐缓冲液(含1%SDS)和pH6.8磷酸盐缓冲液(含1%SDS)为溶出介质,分别于5、10、15、30、45、60、90min时取样用微孔滤膜滤过续滤液作为供试品溶液取20μL进样按外标法以峰面积计算溶出度并计算累积溶出度自制制剂和进口制剂在不同介质中的溶出曲线见图3。经结果比对,采用本发明晶型的甲苯磺酸索拉非尼片,原料过120目筛的自制制剂和原料微粉化的进口制剂的溶出度相似说明自制制剂和参比制剂基本达到了体外溶出一致,说明采用本发明的甲苯磺酸索拉非尼晶型制备的甲苯磺酸索拉非尼口服固体制剂在水中溶出速度快,大大提高了索拉非尼的生物利用度。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1