一种应用于植物上的CRISPR/Cas9载体的构建方法与流程
本发明涉及植物分子生物学领域,尤其涉及应用于植物上的crispr/cas9载体的构建方法。
背景技术:
crispr/cas9技术是自2013年兴起的一种高效简便的基因组编辑技术,目前已在动物、模式植物中得到广泛应用。它主要是基于细菌的一种获得性免疫系统改造而成,由于其可用于对dna进行定点编辑,并且可以同时作用于多个靶位点,同时编辑多个基因,较常规转基因方法具有明显优势,且沉默效果更加彻底,因此越来越多的研究人员对其产生了浓厚兴趣。此外,在常用的基因组编辑技术中,crispr/cas9相对于zfn和talen技术,具有操作简便、制备成本低的巨大优势,使其在常规分子生物学实验室即可使用。
目前,应用于动物上的crispr/cas9载体已经有大量的报道,而应用于植物上的crispr/cas9载体则相对较少,特别是能够进行多位点编辑的载体。
技术实现要素:
为克服现有技术存在的上述技术问题,本发明提供了能够应用于植物上的crispr/cas9载体的构建方法,由其获得的载体不仅能够作用于单个靶位点,而且能够同时作用于两个靶位点。
本发明解决上述技术问题的技术方案如下:一种应用于植物上的crispr/cas9载体的构建方法,其包括:
s1:靶序列退火复性:根据选定的靶序列,合成互补的oligodna,将合成的oligodna序列进行退火复性获得dna双链序列,并稀释;
s2:psg载体的酶切:采用限制性内切酶bbsi酶切psg载体,酶切产物经超薄产物纯化试剂盒进行回收;
s3:连接和转化:配置连接体系,将s1获得的稀释后的dna双链序列与s2获得的酶切产物进行连接反应,将获得的全部连接产物采用热激发转化至大肠杆菌jm109中;
s4:重组质粒的鉴定和提取:分别挑单菌落于lb/amp液体培养基中震荡培养,分别以m13fwd和oligo-r为引物进行菌液pcr鉴定,将验证正确的菌液转接到新鲜的lb/amp液体培养基中,培养后进行质粒的提取,得到重组质粒;
s5:重组资料和pcc质粒的双酶切:将得到的重组质粒和pcc质粒进行双酶切,酶切产物经1%的琼脂糖凝胶电泳后,采用凝胶回收试剂盒分别回收目标片段;
s6:连接、转化和鉴定:配置连接体系,将s5获得的酶切回收目标片段进行连接反应,将获得的全部连接产物采用热激发转化至大肠杆菌jm109中,挑单菌落于lb/kan液体培养基中震荡培养,并进行菌液pcr鉴定,将阳性菌液转接到新鲜的lb/kan液体培养基中培养,提取质粒,即获得构建好的crispr/cas9载体。
在上述技术方案的基础上,本发明还可以做如下改进。
进一步,所述psg载体的构建包括:
以px330质粒为模板,使用高保真酶primestarhsdnapolymerase扩增sgrna片段,回收该片段,标记为sgrna1,引物序列为seqidno.1所示的sg1-f和seqidno.2所示的sg1-r;
使用ecori-hf和xbai分别双酶切puc19和sgrna1,回收目的片段后按1:7的摩尔比进行连接,得到重组质粒psg1,测序,保留序列完全正确的阳性质粒;
以psg1质粒为模板,使用高保真酶primestarhsdnapolymerase扩增sgrna片段,回收该片段,标记为sgrna,引物序列为seqidno.3所示的sg2-f和seqidno.4所示的sg2-r;
使用ecori-hf和xbai双酶切puc19,使用bsai酶切sgrna,回收目的片段后按1:7的摩尔比进行连接,得到重组质粒psg,测序,保留序列完全正确的阳性质粒。
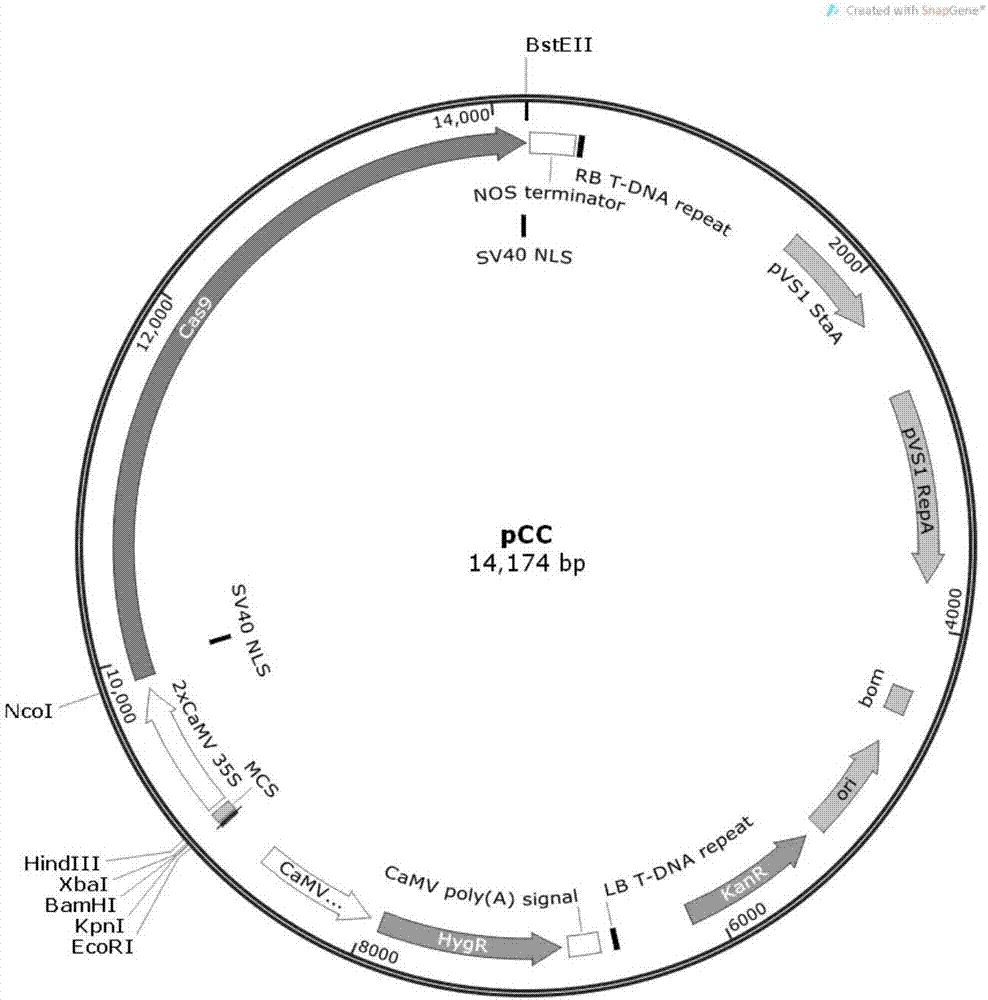
进一步,所述pcc载体的构建包括:
以px330质粒为模板,使用高保真酶primestarhsdnapolymerase扩增hspcas9片段,其中引物cas-f:cas-r1:cas-r2=1.5:0.2:1.3,回收该片段,标记为hspcas9,cas-f的序列如seqidno.5所示,cas-r1的序列如seqidno.6所示,cas-r2的序列如seqidno.7所示;
使用ncoi-hf和bsteii-hf分别双酶切pcambia1302和hspcas9,回收目的片段后按1:5的摩尔比进行连接,得到重组质粒pcc1,测序,保留序列完全正确的阳性质粒;
以pcambia1302质粒为模板,使用高保真酶primestarhsdnapolymerase扩增camv35enhancedpromoter片段,回收该片段,标记为camv-ep,引物序列为seqidno.8所示的camv-ep-f和seqidno.9所示的camv-ep-r;
使用hindiii和ncoi分别双酶切pcc1和camv-ep,回收目的片段后按1:5的摩尔比进行连接,得到重组质粒pcc,测序,保留序列完全正确的阳性质粒。
进一步,在步骤s1中,合成一对互补的oligodna,即序列为seqidno.10所示的oligo-f和序列为seqidno.11所示的oligo-r。
进一步,在步骤s1中,合成两对互补的oligodna,分别为序列为seqidno.12所示的oligo1-f,序列为seqidno.13所示的oligo1-r,序列为seqidno.14所示的oligo2-f及序列为seqidno.15所示的oligo2-r。
进一步,在步骤s1中,将所述合成的oligodna序列进行退火复性的反应程序为:95℃变性5min,每30s降温1℃,降温至25℃,并于4℃保存;在所述步骤s2中,psg载体的酶切的反应体系为100μl,37℃反应过夜,65℃反应20min。
进一步,在所述步骤s4中,所得到的重组质粒为psg-cz;在所述步骤s5中,将得到的psg-cz重组质粒、pcc质粒分别采用ecori-hf和xbai进行双酶切,37℃酶切3h后,65℃反应20min得到所述酶切产物。
进一步,在所述步骤s4中,所得到的重组质粒为psg-cz1和psg-cz2;所述步骤s5中,将得到的psg-cz1重组质粒采用ecori-hf和kpni进行双酶切,psg-cz2重组质粒采用xbai和kpni进行双酶切;或将得到的psg-cz1重组质粒采用ecori-hf和bamhi进行双酶切,psg-cz2重组质粒采用xbai和bamhi进行双酶切;并将pcc质粒采用ecori-hf和xbai进行双酶切,37℃酶切3h后,65℃反应20min,得到所述酶切产物。
进一步,在所述步骤s6中,所述菌液pcr鉴定以m13rev和oligo-r为引物。
进一步,在所述步骤s6中,所述菌液pcr鉴定以oligo1-f和oligo2-r为引物。
与现有技术相比,本发明提供的应用于植物上的crispr/cas9载体的构建方法可获得应用于植物上的crispr/cas9载体,可用于下一步的遗传转化试验,该载体不仅能够作用于单个靶位点,而且能够同时作用于两个靶位点。
附图说明
图1为psg载体的图谱;
图2为pcc载体的图谱;
图3为本发明提供的应用于植物上的crispr/cas9载体的构建方法的流程图。
具体实施方式
以下结合附图对本发明的原理和特征进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。需要说明的是,在不冲突的情况下,本申请的实施例及实施例中的特征可以相互组合。
本发明的目的在于提供一种能够应用于植物上的crispr/cas9载体,该载体不仅能够作用于单个靶位点,而且能够同时作用于两个靶位点。为实现此目标,该crispr/cas9载体由两个基本载体psg和pcc组成,构建方法具体包含以下内容:
1.psg载体的构建:
1)以px330质粒为模板,使用高保真酶primestarhsdnapolymerase扩增sgrna片段,回收该片段,标记为sgrna1,引物序列如下:
(seqidno.1)sg1-f:
ggaattcatagtttcccatgattccttcatatttgc(下划线标记的为ecori酶切位点);
(seqidno.2)sg1-r:
tacctctagagccatttgtctgc(下划线标记的为xbai酶切位点);
2)使用ecori-hf和xbai分别双酶切puc19和sgrna1,回收目的片段后按1:7的摩尔比进行连接,得到重组质粒psg1,测序,保留序列完全正确的阳性质粒;
3)以psg1质粒为模板,使用高保真酶primestarhsdnapolymerase扩增sgrna片段,回收该片段,标记为sgrna,引物序列如下:
(seqidno.3)sg2-f:
atatatggtctcaaattggatccggtaccgaattcatagtttcccatgattcct(下划线标记的为bsai、bamhi、kpni、ecori酶切位点);
(seqidno.4)sg2-r:
atatatggtctcactagggatccggtaccctctagagccatttgtctgcagaatt(下划线标记的为bsai、bamhi、kpni、xbai酶切位点);
4)使用ecori-hf和xbai双酶切puc19,使用bsai酶切sgrna,回收目的片段后按1:7的摩尔比进行连接,得到重组质粒psg,测序,保留序列完全正确的阳性质粒,psg载体的图谱如图1所示,psg载体的部分序列如下所示(seqidno.16):
其中
2.pcc载体的构建
5)以px330质粒为模板,使用高保真酶primestarhsdnapolymerase扩增hspcas9片段,其中引物cas-f:cas-r1:cas-r2=1.5:0.2:1.3,回收该片段,标记为hspcas9,引物序列如下:
(seqidno.5)cas-f:
catgccatggactataaggaccacgacggagact(下划线标记的为ncoi酶切位点);
(seqidno.6)cas-r1:
gaccttccgcttcttctttggctttttcttttttgcctggccggcct;
(seqidno.7)cas-r2:
cagggtcaccttaaccgaccttccgcttcttctttggct(下划线标记的分别为bsteii酶切位点和sv40核定位信号序列);
6)使用ncoi-hf和bsteii-hf分别双酶切pcambia1302和hspcas9,回收目的片段后按1:5的摩尔比进行连接,得到重组质粒pcc1,测序,保留序列完全正确的阳性质粒;
7)以pcambia1302质粒为模板,使用高保真酶primestarhsdnapolymerase扩增camv35enhancedpromoter片段,回收该片段,标记为camv-ep,引物序列如下:
(seqidno.8)camv-ep-f:
cccaagcttttgcgtattggctagagcagcttg(下划线标记的为hindiii酶切位点);
(seqidno.9)camv-ep-r:
catgccatggctcattgccccccgggatct(下划线标记的为ncoi酶切位点);
8)使用hindiii和ncoi分别双酶切pcc1和camv-ep,回收目的片段后按1:5的摩尔比进行连接,得到重组质粒pcc,测序,保留序列完全正确的阳性质粒,pcc载体的图谱如图2所示。
具体地,crispr/cas9载体的构建方法如图3所示,包括:
s1:靶序列退火复性:根据选定的靶序列,合成互补的oligodna,将合成的oligodna序列进行退火复性获得dna双链序列,并稀释;
s2:psg载体的酶切:采用限制性内切酶bbsi酶切psg载体,酶切产物经超薄产物纯化试剂盒进行回收;
s3:连接和转化:配置连接体系,将s1获得的稀释后的dna双链序列与s2获得的酶切产物进行连接反应,将获得的全部连接产物采用热激发转化至大肠杆菌jm109中;
s4:重组质粒的鉴定和提取:分别挑单菌落于lb/amp液体培养基中震荡培养,分别以m13fwd和oligo-r为引物进行菌液pcr鉴定,将验证正确的菌液转接到新鲜的lb/amp液体培养基中,培养后进行质粒的提取,得到重组质粒;
s5:重组资料和pcc质粒的双酶切:将得到的重组质粒和pcc质粒进行双酶切,酶切产物经1%的琼脂糖凝胶电泳后,采用凝胶回收试剂盒分别回收目标片段;
s6:连接、转化和鉴定:配置连接体系,将s5获得的酶切回收目标片段进行连接反应,将获得的全部连接产物采用热激发转化至大肠杆菌jm109中,挑单菌落于lb/kan液体培养基中震荡培养,并进行菌液pcr鉴定,将阳性菌液转接到新鲜的lb/kan液体培养基中培养,提取质粒,即获得构建好的crispr/cas9载体。
实施方式1
本实施方式提供了作用于单个位点的crispr/cas9载体制备过程:
(1)靶序列退火复性。根据选定的靶序列,合成一对互补的oligodna,序列为:oligo-f(seqidno.10):caccnnnnnnnnnnnnnnnnnnnn,oligo-r(seqidno.11):aaacnnnnnnnnnnnnnnnnnnnn。将合成的oligo序列按照表1进行退火复性,反应程序为:95℃变性5min,1℃/30s降温至25℃,4℃保存。将得到的dna双链序列稀释至0.1μm。
表1靶序列退火复性的反应体系
(2)psg质粒的酶切。采用限制性内切酶bbsi酶切psg载体,反应体系100μl,如表2所示,37℃反应过夜,65℃反应20min,酶切产物经超薄产物纯化试剂盒进行回收,并使用核酸蛋白仪测定浓度。
表2bbsi酶切psg载体的反应体系
(3)连接和转化。按照表3配置连接体系,16℃反应30min后4℃反应过夜;将全部连接产物采用热激发转化至大肠杆菌jm109中。
表3复性产物与psg酶切片段的连接反应体系
(4)重组质粒的鉴定和提取。挑单菌落于800μl的lb/amp液体培养基中,37℃振荡培养。以m13fwd和oligo-r为引物进行菌液pcr鉴定,将验证正确的菌液转接到新鲜的lb/amp液体培养基中,培养后进行质粒的提取,得到重组质粒psg-cz。
(5)psg-cz和pcc质粒的双酶切。将得到的psg-cz重组质粒、pcc质粒分别采用ecori-hf和xbai进行双酶切,37℃酶切3h后65℃反应20min;酶切产物经1%的琼脂糖凝胶电泳后,采用凝胶回收试剂盒分别回收目标片段,并用核酸蛋白仪测定浓度。
(6)连接、转化和鉴定。按照表4配置连接体系,16℃反应30min后4℃反应过夜;将全部连接产物采用热激发转化至大肠杆菌jm109中;挑单菌落于800μl的lb/kan液体培养基中,37℃振荡培养;以m13rev和oligo-r为引物进行菌液pcr鉴定,将阳性菌液转接到新鲜的lb/kan液体培养基中培养,提取质粒,-20℃保存。该质粒即为构建好的作用于单个位点的crispr/cas9载体,可用于下一步的遗传转化试验。
表4酶切回收片段的连接反应体系
实施方式2
本实施方式提供了作用于两个位点的crispr/cas9载体制备过程:
(1)靶序列退火复性。根据选定的靶序列,合成两对互补的oligodna,序列为:
oligo1-f(seqidno.12):caccnnnnnnnnnnnnnnnnnnnn;
oligo1-r(seqidno.13):aaacnnnnnnnnnnnnnnnnnnnn;
oligo2-f(seqidno.14):caccnnnnnnnnnnnnnnnnnnnn;
oligo2-r(seqidno.15):aaacnnnnnnnnnnnnnnnnnnnn;
将合成的oligo序列分别按照表5进行退火复性,反应程序为:95℃变性5min,1℃/30s降温至25℃,4℃保存;将得到的dna双链序列稀释至0.1μm。
表5靶序列退火复性的反应体系
(2)psg质粒的酶切。采用限制性内切酶bbsi酶切psg载体,反应体系100μl(表6),37℃反应过夜,65℃反应20min,酶切产物经超薄产物纯化试剂盒进行回收,并使用核酸蛋白仪测定浓度。
表6bbsi酶切psg载体的反应体系
(3)连接和转化。按照表7分别配置连接体系,16℃反应30min后4℃反应过夜。将全部连接产物采用热激发转化至大肠杆菌jm109中。
表7复性产物与psg酶切片段的连接反应体系
(4)重组质粒的鉴定和提取。分别挑单菌落于800μl的lb/amp液体培养基中,37℃振荡培养;分别以m13fwd和oligo-r为引物进行菌液pcr鉴定,将验证正确的菌液转接到新鲜的lb/amp液体培养基中,培养后进行质粒的提取,得到重组质粒psg-cz1和psg-cz2。
(5)psg-cz1、psg-cz2和pcc质粒的双酶切。将得到的psg-cz1重组质粒采用ecori-hf和kpni进行双酶切,psg-cz2重组质粒采用xbai和kpni进行双酶切;或将得到的psg-cz1重组质粒采用ecori-hf和bamhi进行双酶切,psg-cz2重组质粒采用xbai和bamhi进行双酶切;将pcc质粒采用ecori-hf和xbai进行双酶切,37℃酶切3h后65℃反应20min。酶切产物经1%的琼脂糖凝胶电泳后,采用凝胶回收试剂盒分别回收目标片段,并用核酸蛋白仪测定浓度。
(6)连接、转化和鉴定。按照表8配置连接体系,16℃反应30min后4℃反应过夜;将全部连接产物采用热激发转化至大肠杆菌jm109中;挑单菌落于800μl的lb/kan液体培养基中,37℃振荡培养;以oligo1-f和oligo2-r为引物进行菌液pcr鉴定,将阳性菌液转接到新鲜的lb/kan液体培养基中培养,提取质粒,-20℃保存。该质粒即为构建好的作用于两个位点的crispr/cas9载体,可用于下一步的遗传转化试验。
表8酶切回收片段的连接反应体系
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
sequencelisting
<110>四川农业大学
<120>一种应用于植物上的crispr/cas9载体的构建方法
<130>2017
<160>16
<170>patentinversion3.3
<210>1
<211>36
<212>dna
<213>人工序列
<400>1
ggaattcatagtttcccatgattccttcatatttgc36
<210>2
<211>23
<212>dna
<213>人工序列
<400>2
tacctctagagccatttgtctgc23
<210>3
<211>54
<212>dna
<213>人工序列
<400>3
atatatggtctcaaattggatccggtaccgaattcatagtttcccatgattcct54
<210>4
<211>55
<212>dna
<213>人工序列
<400>4
atatatggtctcactagggatccggtaccctctagagccatttgtctgcagaatt55
<210>5
<211>34
<212>dna
<213>人工序列
<400>5
catgccatggactataaggaccacgacggagact34
<210>6
<211>47
<212>dna
<213>人工序列
<400>6
gaccttccgcttcttctttggctttttcttttttgcctggccggcct47
<210>7
<211>39
<212>dna
<213>人工序列
<400>7
cagggtcaccttaaccgaccttccgcttcttctttggct39
<210>8
<211>33
<212>dna
<213>人工序列
<400>8
cccaagcttttgcgtattggctagagcagcttg33
<210>9
<211>30
<212>dna
<213>人工序列
<400>9
catgccatggctcattgccccccgggatct30
<210>10
<211>24
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(24)
<223>nisa,c,g,ort
<400>10
caccnnnnnnnnnnnnnnnnnnnn24
<210>11
<211>24
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(24)
<223>nisa,c,g,ort
<400>11
aaacnnnnnnnnnnnnnnnnnnnn24
<210>12
<211>24
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(24)
<223>nisa,c,g,ort
<400>12
caccnnnnnnnnnnnnnnnnnnnn24
<210>13
<211>24
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(24)
<223>nisa,c,g,ort
<400>13
aaacnnnnnnnnnnnnnnnnnnnn24
<210>14
<211>24
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(24)
<223>nisa,c,g,ort
<400>14
caccnnnnnnnnnnnnnnnnnnnn24
<210>15
<211>24
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(24)
<223>nisa,c,g,ort
<400>15
aaacnnnnnnnnnnnnnnnnnnnn24
<210>16
<211>568
<212>dna
<213>人工序列
<400>16
acgacgttgtaaaacgacggccagtgaattggatccggtaccgaattcatagtttcccat60
gattccttcatatttgcatatacgatacaaggctgttagagagataattggaattaattt120
gactgtaaacacaaagatattagtacaaaatacgtgacgtagaaagtaataatttcttgg180
gtagtttgcagttttaaaattatgttttaaaatggactatcatatgcttaccgtaacttg240
aaagtatttcgatttcttggctttatatatcttgtggaaaggacgaaacaccgggtcttc300
gagaagacctgttttagagctagaaatagcaagttaaaataaggctagtccgttatcaac360
ttgaaaaagtggcaccgagtcggtgcttttttgttttagagctagaaatagcaagttaaa420
ataaggctagtccgtttttagcgcgtgcgccaattctgcagacaaatggctctagagggt480
accggatccctagagattaatcgtcgacctgcaggcatgcaagcttggcgtaatcatggt540
catagctgtttcctgtgtgaaattgtta568
- 还没有人留言评论。精彩留言会获得点赞!