一种Bola型三氮唑核苷化合物及其制备方法和应用与流程
本发明属于化学药物领域,涉及一种Bola型三氮唑核苷化合物,还涉及其制备方法和应用。
背景技术:
核苷类似物是一类重要的抗病毒和抗癌药物。它们具有与天然核苷类似的结构,并能够模拟天然核苷,参与病毒和癌细胞的DNA/RNA合成,或与病毒和癌细胞中某些酶相互作用,从而达到抑制病毒复制和癌细胞生长的目的。人们通过对核苷的核糖部分或者碱基部分进行修饰,得到了各种具有抗病毒和抗癌活性的核苷药物,如利巴韦林(ribavirin)、拉米夫定(Lamivudine)、吉西他滨(gemcitabine)、5-氟尿苷(floxuridine)和阿糖胞苷(cytarabine)等。其中利巴韦林作为核苷类药物的杰出代表,具有广谱抗病毒性质,对多种DNA病毒和RNA病毒都有很好的抑制作用,是目前治疗丙型肝炎病毒感染(Hepatitis C Virus,HCV)的唯一小分子药物,同时在近期也被证实可作为抗癌药物治疗急性骨髓性白血病。和常见的核苷类似物不同,利巴韦林是以非天然的杂环-1,2,4-三氮唑取代了天然核苷的碱基部分,这种特殊结构的优势在于不易被体内的核苷/核酸代谢酶识别,因而该类化合物在体内有更好的代谢稳定性,同时三氮唑作为一种通用的碱基,有着介于嘌呤和嘧啶碱之间的特殊几何构型以及广泛的氢键结合能力。另外值得一提的是以利巴韦林为例,三氮唑核苷不仅具有抗病毒活性,同时也能调节生物体系的免疫反应,这为我们发展出同时具有抗代谢和调节免疫反应的药物先导用于抗癌和抗病毒治疗提供了研究基础。近年来,为了进一步增加该类分子的芳基结合平面,从而提高其与生物靶标分子的结合能力,研究者们尝试向三氮唑核苷的碱基上引入芳香基团,得到新的芳基三氮唑核苷类似物并将其作为药物先导广泛应用于各种病毒和癌症相关疾病的治疗。但是和大多数核苷类似物一样,三氮唑核苷化合物也面临水溶性差和起效剂量大的问题,使得其进入临床研究的可能性受到影响,这也是目前发展该类化合物作为药物先导中一个待以解决的难题。另外,作为小分子药物,在癌症治疗中对正常组织和肿瘤缺乏选择性,难以真正作用于病变部位也是制约该类药物疗效,并导致药物毒副作用的根本原因。因此发展水溶性的新型三氮唑核苷衍生物,并基于其结构通过小分子自组装将其转化为纳米药物是改善这类化合物的物理化学性质,并增强其靶向病灶组织的能力,从而减少对正常组织和细胞伤害的重要途径。
Bola型的两亲分子是将两个极性头基用疏水链连接起来,这种特殊结构赋予了Bola型分子独特的性质,如在质膜中具有更好的稳定性和兼容性等。目前Bola型两亲分子被广泛应用于表面活性剂,其自组装形成的囊泡结构也被作为一种理想的载体用于药物传递。
技术实现要素:
有鉴于此,本发明的目的在于提供一种Bola型三氮唑核苷化合物及其制备方法和应用。
为达到上述目的,本发明提供如下技术方案:
一种Bola型三氮唑核苷化合物,结构如通式I所示:
式中:
R1为其中R5和R6各自独立为-H,-OH,-NH2,-NHCH3,-N(CH3)2或氨基酸其中的一种,R7为-OH,-NH2,-NHCH3,-N(CH3)2或氨基酸其中的一种;
R2为-H,-Cl,-Br,-I或-Ar;
R3为-CH2-或-O-;
R4为-CH2-,-S-S-,
n独立地表示等于2~10的整数。
如通式I化合物的纳米颗粒,取通式I化合物加水,超声溶解后用0.45μm滤膜过滤得Bola型三氮唑核苷化合物纳米颗粒。
如通式I化合物的制备方法:按摩尔比为1:2~10分别取通式II化合物和通式III化合物反应得到通式I所示Bola型三氮唑核苷化合物;
通式II所示化合物中X为-CH2-,-S-S-,Y为炔基或羟基;n为2~10整数;
通式III所示化合物中R1为其中R5和R6各自独立为-H,-OH,-NH2,-NHCH3,-N(CH3)2或氨基酸其中的一种,R7为-OH,-NH2,-NHCH3,-N(CH3)2或氨基酸其中的一种;R2为-H,-Cl,-Br,-I或-Ar;R8为-N3或-Br。
进一步,当通式II中Y为炔基,通式III中R8为叠氮基时,如通式1化合物制备方法为:按摩尔比为1:2~10分别取通式II化合物和通式III化合物混合后用体积比为1:1~5四氢呋喃和水的混合溶剂溶解,再加入抗坏血酸钠溶液和五水硫酸铜溶液,80℃下反应0.5~6小时,旋干溶剂,柱层析分离得通式I所示Bola型三氮唑核苷化合物。
进一步,当通式II中Y为炔基,通式III中R8为-Br时,如通式1化合物制备方法为:按摩尔比为1:2~10分别取通式II化合物和通式III化合物混合后用体积比为1:1~5二氧六环和水的混合溶剂溶解,再加入四三苯基膦钯、碘化亚铜和碳酸锂,100℃微波反应15~60分钟,旋干溶剂后柱层析分离,分离产物在氢气作用下还原得到通式I所示Bola型三氮唑核苷化合物,所述四三苯基膦钯、碘化亚铜、碳酸锂和通式III化合物的摩尔比为1:1:40:20。
进一步,当通式II中Y为羟基,通式III中R8为-Br时,如通式I化合物制备方法为:按摩尔比为1:2~10分别取通式II化合物和通式III化合物混合后用二甲基甲酰胺作溶剂,再加入Cs2CO3,搅拌8~24小时,旋干溶剂柱层析得到通式I所示Bola型三氮唑核苷化合物,所述Cs2CO3和通式III化合物的摩尔比为3:1。
进一步,所述通式II化合物碳链上的H原子可替换为F原子。
通式I所示Bola型三氮唑核苷化合物或通式I所示Bola型三氮唑核苷化合物纳米颗粒在抑制细胞生长药物的应用。
进一步,所述细胞为癌细胞。
进一步,所述癌细胞为前列腺癌细胞PC-3、肝癌细胞HepG2、宫颈癌细胞Hela、卵巢癌细胞SKOV3、胰腺癌细胞BxPC-3和Panc-1。
本发明的有益效果在于:基于三氮唑核苷利巴韦林进行结构优化所制备的bola型三氮唑核苷化合物均具有抑制癌细胞增殖的活性。且在相同浓度下,这一类化合物的抑制癌细胞增殖的活性均优于利巴韦林,其中化合物I-1对所有癌细胞增殖的抑制效果都远优于相应的临床用药。化合物I-2在相同浓度下与临床用药多西紫杉醇对前列腺癌PC-3细胞的抑制效果相当,比肝癌临床用药五氟尿嘧啶对HepG2细胞的抑制效果要好,比宫颈癌临床用药顺铂对Hela细胞的抑制效果要好,也比胰腺癌临床用药吉西他滨对BxPC-3和Panc-1细胞的抑制效果要好。化合物I-3在相同浓度下与临床用药多西紫杉醇对前列腺癌PC-3细胞的抑制效果相当,比肝癌临床用药五氟尿嘧啶对HepG2细胞的抑制效果要好,也比胰腺癌临床用药吉西他滨对BxPC-3和Panc-1细胞的抑制效果要好。因此本发明提供一类新型的抗肿瘤药物先导。同时这类化合物有在水溶液中自组装形成纳米尺度颗粒的能力,也使其能因为实体瘤的高通透性和滞留效应(Enhanced Permeability and Retention effect,EPR)更易聚集在肿瘤组织内部实现靶向给药,同时作为纳米药物载体还可以包封其他小分子药物而达到联合用药的效果。
附图说明
为了使本发明的目的、技术方案和有益效果更加清楚,本发明提供如下附图进行说明:
图1为动态光散射表征本发明化合物I-1自组装形成的纳米尺度的颗粒;
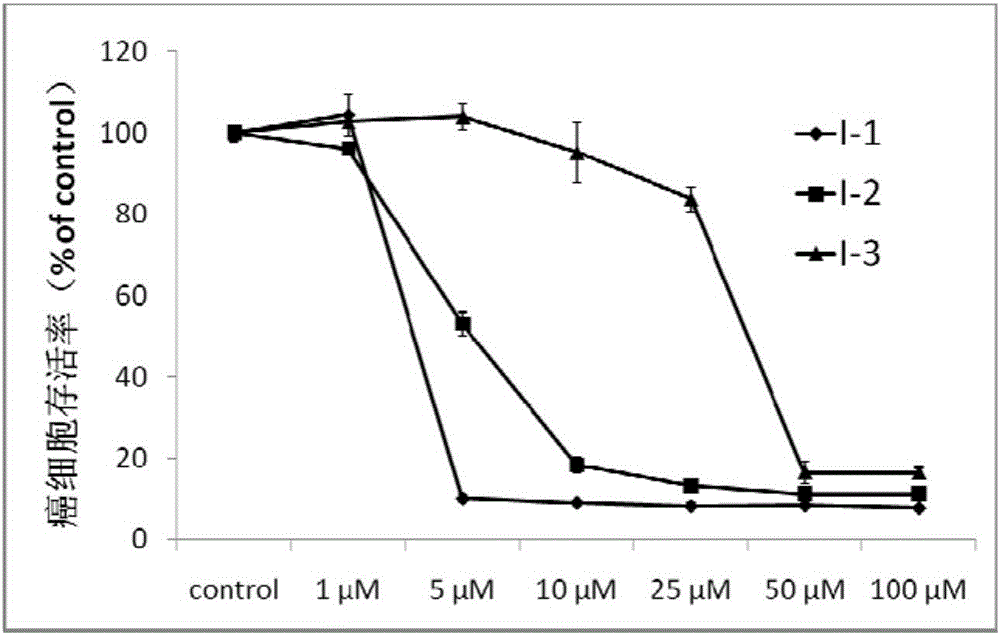
图2为本发明化合物I-1~I-3随浓度升高对前列腺癌PC-3细胞增殖的抑制能力;
图3为本发明化合物I-1~I-3随浓度升高对肝癌细胞HepG2增殖的抑制能力;
图4为本发明化合物I-1~I-3随浓度升高对胰腺癌细胞Panc-1增殖的抑制能力;
图5为本发明化合物I-1和I-2随浓度升高对胰腺癌细胞BxPC-3增殖的抑制能力;
图6为本发明化合物I-1和I-2随浓度升高对宫颈癌Hela细胞增殖的抑制能力;
图7为本发明化合物I-1随浓度升高对卵巢癌SKOV3细胞增殖的抑制能力。
具体实施方式
下面将结合附图,对本发明的优选实施例进行详细的描述。
实施例1
反应中间体II-1的制备
将3.3mL II-1a(1,8-二溴辛烷)溶解在3mL二甲基甲酰胺中,滴加含1.6g乙炔钠的二甲苯溶液,反应24小时后过滤反应液,旋干溶剂,减压蒸馏,得产物II-1b。在圆底烧瓶中分别加入630mg化合物II-1b和660mg硫代乙酸钾,加DMF作溶剂,室温下反应4小时后旋干溶剂,加入二氯甲烷和水后分离有机层,旋干溶剂后经柱层析分离,得化合物II-1c。将100mg化合物A3溶解于DMF,室温下滴加80μL水合肼,再滴加100μL乙酸,室温下反应30分钟后加入水和乙酸乙酯萃取,有机相经无水硫酸钠干燥,旋干溶剂,得化合物II-1d。II-1d与2,6-二氟苯甲醛混合后加入二氯甲烷溶解,再在反应体系中滴加三氟化硼的乙醚溶液,0℃下反应1小时后,反应液用饱和碳酸氢钠水溶液洗涤,旋干溶剂后分离得到化合物II-1。
II-1:1H NMR(400MHz,CDCl3):δ7.23-7.18(m,1H),6.89(t,J=8Hz,2H),5.15(s,1H),2.63(t,J=8Hz,4H),2.19-2.15(m,4H),1.94(s,2H),1.62-1.56(m,4H),1.54-1.47(m,4H),1.43-1.27(m,16H).
实施例2
反应中间体II-2的制备
将75mg化合物II-1b和9.5mg磷酸钾溶解在5mL乙腈中,通入氧气,室温下反应5h。旋干溶剂,柱层析分离,得化合物II-2,产率77.8%。
II-2:1H NMR(400MHz,CDCl3):δ2.68(t,J=8Hz,4H),2.18(t,J=4Hz,4H),1.94(s,2H),1.71-1.63(m,4H),1.56-1.50(m,4H),1.39-1.31(m,16H).
实施例3
反应中间体II-3的制备
将5.1mL三甲基硅基乙炔溶解在45mL四氢呋喃中,在-78℃下滴加23.2mL丁基锂。15分钟后,加入28mL DMPU,在-78℃下搅拌1小时。随后加入2.8g化合物II-3a的四氢呋喃溶液2mL,在室温下反应过夜。反应液用1N的盐酸调节pH值至1-2,并用乙酸乙酯萃取3次。合并有机相,无水硫酸钠干燥,旋干溶剂,柱层析分离得化合物II-3b。将1.6g化合物II-3b溶解在TBAF的四氢呋喃溶液中,在室温下反应3h。用氯化铵溶液淬灭反应,并用乙酸乙酯萃取3次,合并有机相,无水硫酸钠干燥,旋干溶剂,柱层析分离得化合物II-3c。将1.0g化合物II-3c溶解在2mL四氢呋喃中,在0℃下滴加1.13g氢化锂铝的四氢呋喃悬浮液30mL,在室温下反应3h。用氯化铵溶液淬灭反应,过滤,浓缩滤液,柱层析分离得化合物II-3d-1。另将将3.84g PCC和0.145g乙酸钠溶解在15mL二氯甲烷中,在0℃下加入1.5g化合物II-3d-1,在室温下反应6小时。旋干溶剂,柱层析分离得化合物II-3e-1。将1.0g化合物II-3c溶解在8.6mL乙醇中,加入0.1mL浓硫酸,回流反应6小时。反应液用乙醚萃取3次,合并有机相,旋干溶剂,柱层析分离得化合物II-3d-2。将168mg化合物II-3d-2和1.1mmol水合肼溶解在50mL乙醇中,回流反应6小时。冷却至室温,放置过夜,收集固体,用乙醇重结晶得化合物II-3e-2。将138mg化合物II-3e-1和184mg化合物II-3e-2溶解在5mL乙醇中,加入0.2mL乙酸,回流反应2小时。柱层析分离得化合物II-3。
实施例4
反应中间体III-1的制备
在圆底烧瓶中加入86mg化合物III-1a,360mg对甲苯磺酰氯,6mg 4-二甲氨基吡啶和0.3mL三乙胺,然后加入二氯甲烷使之溶解。室温下反应3小时,旋干溶剂后柱层析分离,得化合物III-1b。140mg化合物III-1b和360mg溴化锂混合后,加入丙酮使之溶解。回流反应3小时后,旋干溶剂,柱层析分离得化合物III-1c。100mg化合物III-1c和450mg二甲胺水溶液混合后,再加入DMF使之溶解。在室温下反过夜,旋干溶剂后分离得到化合物III-1。III-1:1H NMR(400MHz,DMSO-d6):δ8.71(s,1H),5.46(s,2H),3.58(t,J=4Hz,2H),2.46(t,J=4Hz,2H),2.18(s,6H).
实施例5
反应中间体III-2的制备
将600mg化合物III-2a溶解在4mL含有216mg NaOH的水溶液中,加入0.3mL液溴,缓慢升温至100℃,保温反应20小时。冷却至室温,用饱和NaHSO3溶液淬灭,用乙醚萃取后合并有机相,无水硫酸钠干燥。旋干溶剂,得877mg化合物III-2b。在圆底烧瓶中分别加入190mg化合物III-2b,440mg乙酰基保护的2-(羟基甲氧基)乙醇,2mg水合对甲苯磺酸,140℃下反应1h。柱层析分离,得化合物III-2c。在980mg的化合物III-2c中加入25mL饱和氨的甲醇溶液,室温下反应48小时。旋干溶剂,柱层析分离,得化合物化合物III-2。
III-2:1H NMR(400MHz,CDCl3):δ5.48(s,2H),3.75(t,J=4Hz,2H),3.73(t,J=4Hz,2H).
实施例6
反应中间体III-3的制备
将200mg化合物III-1溶解在5mL甲醇中,在钯碳/氢气的条件下,搅拌2小时,过滤,旋干溶剂,得到化合物III-3a。将100mg化合物III-3a溶解在5mL的20%硫酸溶液中,在0℃下滴加含有37mg NaNO2的水溶液0.4mL,0℃下反应30分钟后,再滴加含有80mg CuBr的水溶液,室温下反应2小时。然后用乙酸乙酯萃取并合并有机相,无水硫酸钠干燥以后,旋干溶剂,柱层析分离得化合物III-3。
实施例7
化合物I-1的制备
65mg化合物II-1和60mg化合物III-1混合后用体积比为1:2的四氢呋喃和水的混合溶剂溶解,再加入抗坏血酸钠溶液0.2mL和五水硫酸铜溶液0.2mL,80℃下反应2小时。旋干溶剂,柱层析分离,得化合物82mg化合物I-1。
I-1:1H NMR(400MHz,DMSO-d6):δ8.94(s,2H),8.43(s,2H),7.42-7.35(m,1H),7.10(t,J=8Hz,2H),5.61(s,4H),5.17(s,1H),3.63(t,J=4Hz,4H),3.69(t,J=8Hz,4H),2.65-2.55(m,4H),2.50(s,12H),2.39(t,J=4Hz,4H),1.67-1.60(m,4H),1.54-1.47(m,4H),1.29-1.24(m,16H);13C NMR(100MHz,DMSO):δ155.60,148.17,147.14,130.43,121.48,118.11,112.56,112.35,78.69,67.77,58.31,45.83,42.15,33.19,29.09,29.03,28.98,28.86,28.81,28.40,25.10;MS(ESI)m/z:887.64[M+H].
实施例8
化合物I-2的制备
120mg化合物II-1和136mg化合物III-2混合后用体积比为1:2的四氢呋喃和水的混合溶剂溶解,再加入抗坏血酸钠溶液0.2mL和五水硫酸铜溶液0.2mL,80℃下反应2小时。旋干溶剂,柱层析分离得146mg化合物I-2。
I-2:1H NMR(400MHz,CDCl3):δ7.99(s,2H),7.23-7.17(m,1H),6.89(t,J=8Hz,2H),5.65(s,4H),5.15(s,1H),4.74(t,J=4Hz,2H),3.82-3.79(m,8H),2.79(t,J=8Hz,4H),2.63(t,J=8Hz,4H),2.07(s,2H),1.74-1.66(m,4H),1.61-1.55(m,4H),1.37-1.29(m,16H);13C NMR(100MHz,DMSO):δ155.32,148.36,132.14,130.44,121.48,118.11,112.56,112.36,79.10,71.88,60.25,42.16,33.20,29.09,29.03,28.98,28.84,28.81,28.41,25.08;MS(ESI):m/z 989.36[M+H].
实施例9
化合物I-3的制备
在圆底烧瓶中分别加入20mg II-1和15.8mg的III-4,随后用体积比为1:2的四氢呋喃和水的混合溶剂溶解。再加入新制的抗坏血酸钠溶液0.1mL和五水硫酸铜溶液0.1mL,80℃下反应2小时。旋干溶剂,柱层析分离,得25mg化合物I-3。
I-3:1H NMR(400MHz,DMSO-d6):δ8.94(s,2H),8.43(s,2H),7.42-7.35(m,1H),7.11(t,J=8Hz,2H),5.63(s,4H),5.17(s,1H),4.72(t,J=4Hz,2H),3.59(t,J=4Hz,4H),3.51(t,J=8Hz,4H),2.69(t,J=8Hz,4H),2.64-2.55(m,4H),1.67-1.62(m,4H),1.54-1.47(m,4H),1.29-1.24(m,16H).实施例10
化合物I-4的制备
在圆底烧瓶中分别加入76mg化合物III-1,58mg化合物II-2,随后加入体积比为1:2的四氢呋喃和水的混合溶剂使之溶解。加入新制的抗坏血酸钠溶液0.2mL和五水硫酸铜溶液0.2mL,80℃下反应2小时。旋干溶剂,柱层析分离,得76.5mg化合物I-4。
I-4:1H NMR(400MHz,DMSO-d6):δ8.94(s,2H),8.43(s,2H),5.61(s,4H),3.63(t,J=4Hz,4H),2.72-2.66(m,8H),2.39(t,J=4Hz,4H),2.11(s,12H),1.67-1.57(m,8H),1.30-1.24(m,16H).
实施例11
化合物I-5的制备
在圆底烧瓶中分别加入37mg化合物III-1,60mg化合物II-3,随后加入体积比为1:2的四氢呋喃和水的混合溶剂使之溶解。加入新制的抗坏血酸钠溶液0.2mL和五水硫酸铜溶液0.2mL,80℃下反应2小时。旋干溶剂,柱层析分离,得68mg化合物I-5。
实施例12
化合物I-6的制备
取1,9-癸二炔60mg,化合物III-5 200mg加入微波反应瓶用体积比为3:1的二氧六环和水的混合溶剂溶解,再加入四三苯基膦钯51.6mg,氮化亚铜8.6mg及碳酸锂197mg,100℃下微波反应1小时后,旋干溶剂,柱色谱层析分离得到104mg产物I-6a。60mg化合物I-6a在氢气/钯碳条件下,还原得到化合物I-6 54.8mg.
实施例13
化合物I-7的制备
取1,9-壬二醇50mg,加入碳酸铯610mg以及化合物III-3 156mg,以二甲基甲酰胺作溶剂,室温下搅拌反应16小时,除去溶剂后柱层析得到57mg化合物I-6。
实施例14
化合物I-8的制备
在圆底烧瓶中分别加入42mg化合物III-2,40mg化合物II-1c,随后加入体积比为1:2的四氢呋喃和水的混合溶剂使之溶解。再加入新制的抗坏血酸钠溶液0.2mL和五水硫酸铜溶液0.2mL以后,80℃下反应2小时。旋干溶剂,柱层析分离,得到57mg化合物I-8。
I-8:1H NMR(400MHz,DMSO-d6):8.49(s,1H),5.62(s,2H),4.75(t,J=4Hz,1H),3.62(t,J=4Hz,2H),3.51(t,J=4Hz,2H),2.81(t,J=4Hz,2H),2.70(t,J=8Hz,2H),2.31(s,3H),1.67-1.63(m,2H),1.50-1.47(m,2H),1.28-1.23(m,8H).
实施例15
化合物I-1通过自组装形成纳米颗粒。
实施方法:取2mg化合物1加水10ml,超声溶解后用0.45μM滤膜过滤后,在水溶液中观察到丁达尔现象,用动态光散射仪测量本发明化合物I-1自组装形成的纳米尺度的颗粒结果如图1所示,结果显示该化合物在水溶液自组装形成的颗粒的平均粒径是46nm。
实施例16
本发明化合物I-1用于抗癌活性实验
前列腺癌细胞PC-3用含10%FBS的F12K培养基培养,肝癌细胞HepG2和宫颈癌细胞Hela用含10%FBS的DMEM培养基培养,卵巢癌细胞SKOV3用含10%FBS的McCoy's 5A培养基培养,胰腺癌细胞BxPC-3和Panc-1用含10%FBS的RPMI1640培养基培养,然后在96孔观察板上均以10000每孔的密度种植上述细胞。细胞经过24h增殖后,加入不同浓度的待测化合物,以未加任何药物为负对照,利巴韦林为以上所有细胞的正对照,同时PC-3选取多西紫杉醇作为正对照,HepG2选取五氟尿嘧啶为正对照,Hela和SKOV3选取顺铂为正对照,BxPC-3和Panc-1选取吉西他滨为正对照。在37℃以及5%CO2条件下经过48小时培养后,用比色法测定存活的细胞数目(染色剂为3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide,MTT,又称噻唑蓝)。化合物I-1及上述正对照药物在同样浓度(5μM)下对各种癌细胞的抑制率如表1所示,化合物I-1随浓度升高对前列腺癌PC-3细胞增殖的抑制能力如图2所示;化合物I-1随浓度升高对肝癌细胞HepG2增殖的抑制能力如图3所示;化合物I-1随浓度升高对胰腺癌细胞Panc-1增殖的抑制能力如图4所示;化合物I-1随浓度升高对胰腺癌细胞BxPC-3增殖的抑制能力如图5所示;化合物I-1随浓度升高对宫颈癌Hela细胞增殖的抑制能力如图6所示;化合物I-1随浓度升高对卵巢癌SKOV3细胞增殖的抑制能力如图7所示。
表1化合物I-1及各种抗癌临床用药的浓度一致条件下对不同癌细胞的抑制率
NA:no activity
实施例17
实验例2本发明化合物I-2用于抗癌活性实验
前列腺癌细胞PC-3用含10%FBS的F12K培养基培养,肝癌细胞HepG2和宫颈癌细胞Hela用含10%FBS的DMEM培养基培养,胰腺癌细胞BxPC-3和Panc-1用含10%FB S的RPMI1640培养基培养,然后在96孔观察板上均以10000每孔的密度种植上述细胞。细胞经过24h增殖后,加入不同浓度的待测化合物,以未加任何药物为负对照,利巴韦林为以上所有细胞的正对照,同时PC-3选取多西紫杉醇作为正对照,HepG2选取五氟尿嘧啶为正对照,Hela选取顺铂为正对照,BxPC-3和Panc-1选取吉西他滨为正对照。在37℃以及5%CO2条件下经过48小时培养后,用比色法测定存活的细胞数目(染色剂为3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide,MTT,又称噻唑蓝)。化合物I-2及上述正对照药物在同样浓度下(25μM)对各种癌细胞的抑制率如表2所示,化合物I-2随浓度升高对前列腺癌PC-3细胞增殖的抑制能力如图2所示;化合物I-2随浓度升高对肝癌细胞HepG2增殖的抑制能力如图3所示;化合物I-2随浓度升高对胰腺癌细胞Panc-1增殖的抑制能力如图4所示;化合物I-2随浓度升高对胰腺癌细胞BxPC-3增殖的抑制能力如图5所示;化合物I-2随浓度升高对宫颈癌Hela细胞增殖的抑制能力如图6所示。
表2化合物I-2及各种抗癌临床用药的浓度一致条件下对不同癌细胞的抑制率
实施例18
实验例3本发明化合物I-3用于抗癌活性实验
前列腺癌细胞PC-3用含10%FBS的F12K培养基培养,肝癌细胞HepG2用含10%FBS的DMEM培养基培养,胰腺癌细胞BxPC-3和Panc-1用含10%FBS的RPMI1640培养基培养,然后在96孔观察板上均以10000每孔的密度种植上述细胞。细胞经过24h增殖后,加入不同浓度的待测化合物,以未加任何药物为负对照,利巴韦林为以上所有细胞的正对照,同时PC-3选取多西紫杉醇作为正对照,HepG2选取五氟尿嘧啶为正对照,BxPC-3和Panc-1选取吉西他滨为正对照。在37℃以及5%CO2条件下经过48小时培养后,用比色法测定存活的细胞数目(染色剂为3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide,MTT,又称噻唑蓝)。化合物I-3及上述正对照药物在同样浓度下(50μM)对各种癌细胞的抑制率如表3所示,化合物I-3随浓度升高对前列腺癌PC-3细胞增殖的抑制能力如图2所示;化合物I-3随浓度升高对肝癌细胞HepG2增殖的抑制能力如图3所示;化合物I-3随浓度升高对胰腺癌细胞Panc-1增殖的抑制能力如图4所示。
表3化合物I-3及各种抗癌临床用药的浓度一致条件下对不同癌细胞的抑制率
实施例19
实验例4本发明化合物I-8用于抗癌活性实验
前列腺癌细胞PC-3用含10%FBS的F12K培养基培养,肝癌细胞HepG2用含10%FBS的DMEM培养基培养,卵巢癌细胞SKOV3用含10%FBS的McCoy's 5A培养基培养,胰腺癌细胞BxPC-3和Panc-1用含10%FBS的RPMI1640培养基培养,然后在96孔观察板上均以10000每孔的密度种植上述细胞。细胞经过24h增殖后,加入不同浓度的待测化合物,以未加任何药物为负对照,利巴韦林为以上所有细胞的正对照,同时PC-3选取多西紫杉醇作为正对照,HepG2选取五氟尿嘧啶为正对照,SKOV3选取顺铂为正对照,BxPC-3和Panc-1选取吉西他滨为正对照。在37℃以及5%CO2条件下经过48小时培养后,用比色法测定存活的细胞数目(染色剂为3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide,MTT,又称噻唑蓝)。化合物I-8及上述正对照药物在同样浓度下(50μM)对各种癌细胞的抑制率如表4所示。
表4化合物I-8及各种抗癌临床用药的浓度一致条件下对不同癌细胞的抑制率
NA:no activity
综上,从表1-4的结果可以看出,本发明所制备的bola型三氮唑核苷化合物I-1~I-3均具有明显的抗癌细胞增殖的活性,在同样浓度条件下,对肿瘤细胞生长的抑制效果要优于利巴韦林,同时也比部分抗癌临床用药的活性要好,因此是一类极有潜力的抗肿瘤药物先导。而非bola型的化合物I-8除了在BxPC-3上有一定的抑制癌细胞生长的活性,在其他的细胞系上均无明显的抑制活性,进一步说明了Bola型结构对提高该类化合物生物活性的重要性。
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!