一种非ATP竞争性FGFR1抑制剂及其应用的制作方法
本发明属药学领域,具体而言,本发明涉及小分子化合物能够通过非atp依赖的方式抑制fgfr1及其信号通路的激活,从而发挥良好的体内外抗肿瘤效果。
背景技术:
fgfr1,即成纤维细胞生长因子受体1,是成纤维细胞生长因子(fgf)家族的一员,隶属于酪氨酸受体激酶。大量研究证明fgfr1与多种肿瘤密切相关,如胃癌,在胃癌中,fgfr1的表达量与10年生存率及肿瘤远处转移相关。如肺癌,研究表明,fgfr1的表达量可作为非小细胞肺癌的预后标志。如乳腺癌,染色体8p11-12区域(及fgfr1所在区域)的扩增可出现的10%的病人中。此外,前列腺癌、直肠癌、胰腺癌、口腔鳞状细胞癌及肾细胞癌等多种肿瘤中均有fgfr1扩增的现象。因此,靶向fgfr1的抗肿瘤靶向治疗一直是近年来靶向药物研究的热点,但目前尚未有fgfr1抑制剂上市。
到目前为止,大部分的fgfr1抑制剂都是通过竞争atp的结合位点来发挥抑制fgfr1的作用的,称之为竞争性抑制剂。人基因组编码的大约518种酪氨酸受体激酶均公用高度保守的atp结合域,这一特性导致竞争性抑制剂的靶向选择性较差,具有较大的毒副作用。鉴于此,人们开始思考能否不通过竞争atp的结合位点发挥抑制激酶的作用,即非atp竞争性抑制剂。第一个靶向fgfr1的非atp竞争性抑制剂是arq069,随后又有几篇研究报道了非atp竞争性fgfr1抑制剂的抗肿瘤活性,对比atp竞争性抑制剂,非atp竞争性抑制剂确实具有更佳的靶向选择性。
本发明人经过长期和艰苦的研究实践,借鉴之前非atp竞争性fgfr1抑制剂的结构,设计合成新的非atp竞争性fgfr1抑制剂,它们具有抗肿瘤的药理活性。
技术实现要素:
本发明目的在于提供一种非atp竞争性fgfr1抑制剂及其在制备抗肿瘤药物及与肿瘤相关疾病的治疗药物中的应用。
本发明的另一目的是提供一种用于治疗肿瘤的药物组合物,其含有治疗有效量的作为活性成分的非atp竞争性fgfr1抑制剂及其药用辅料。
具体而言,本发明所述非atp竞争性fgfr1抑制剂为如下所述结构的化合物或其可药用盐:
l16h50的分子式为c24h16cl2o2,化学名称为:2e,2'e)-3,3'-(1,4-phenylene)bis(1-(2-chlorophenyl)prop-2-en-1-one)。
鉴于fgfr1在肿瘤中的重要性以及非atp竞争性抑制剂的优点,非atp竞争性fgfr1抑制剂的研究渐渐成为新型fgfr1抑制剂研究的重要方向之一。我们课题组前期研究中,曾报道过多个非atp竞争性fgfr1抑制剂,包括a114、a117、aea4、aea25和l6123等。本发明中设计合成了一个新型的小分子fgfr1激酶抑制剂化合物l16h50,首先用激酶抑制试验检测了l16h50对fgfr1激酶的抑制活性,发现其对fgfr1激酶具有较好的抑制活性,其对激酶抑制活性的ic50为:3.8±2.7μm(详情见实施例2)。用人工构建高表达fgfr1的hek293细胞,证明了l16h50能剂量依赖地抑制fgf2诱导的fgfr1激活(详情见实施例4),在胃癌细胞系sgc7901中验证了l16h50能剂量依赖地抑制fgf2诱导的fgfr1激活,并能抑制fgf2诱导的fgfr1下游蛋白(包括frs2α,akt,erk1/2及plcγ)的磷酸化(详情见实施例4)。atp竞争性实验可以用来检测激酶抑制剂对激酶活性的抑制作用是否会受atp的浓度改变所影响,结果发现随着atp的浓度升高,l16h50对fgfr1激酶的抑制作用几乎不变(详情见实施例3),因此该抑制剂为一种新型的非atp竞争性fgfr1激酶抑制剂。鉴于l16h50对fgfr1较好的抑制活性,进一步用分子对接的方法研究了l16h50与fgfr1的结合位点,结果表明l16h50能够与fgfr1的ala-564、ser-565、gly-567及asp-641氨基酸残基形成较稳定的氢键(详情见实施例5)。因此,本发明提供了一种新型的非atp竞争性的fgfr1激酶抑制剂。
fgfr1信号通路具有多种生物学功能,包括促进细胞生长、胚胎发育、创口愈合等。肿瘤细胞内fgfr1异常高表达,会导致其介导的信号通路异常激活,其与肿瘤细胞的增殖、迁移、生存及血管形成等相关。大量研究表明fgfr1抑制剂在多种肿瘤中具有较好的抗肿瘤效果。因此,我们首先用mtt的方法检测了l16h50对胃癌细胞生长的影响,l16h50能够显著地抑制胃癌细胞的生长,对sgc7901、bgc823及kato-iii这三株胃癌细胞的ic50分别是0.5±0.4μm、1.4±0.2μm和1.6±1.0μm(详情见实施例4),集落形成实验的结果也表明l16h50能够显著地抑制sgc7901、bgc823及kato-iii细胞的集落形成能力(详情见实施例7)。进一步研究了l16h50抑制胃癌细胞生长的机制,流式细胞术的结果表明l16h50能够将细胞周期阻滞在g2/m期,能明显下调细胞中与g2/m期相关的周期蛋白cyclinb1和cdc2p34蛋白的水平(详情见实施例8)。同时用流式细胞术和westernblot检测了l16h50对胃癌细胞凋亡的影响,结果表明l16h50显著地诱导了胃癌细胞的凋亡,上调促凋亡蛋白cleaved-parp的水平并下调了抗凋亡蛋白bcl-2的水平,而caspase-3/caspase-9检测试剂盒的结果表明l16h50能够增加caspase-3/caspase-9的活性(详情见实施例9)。裸小鼠荷sgc7901异位瘤模型显示,l16h50在动物体内能显著地抑制肿瘤的生长以及抑制fgfr1及其下游蛋白的激活(详情见实施例10)。用急性毒性实验评估l16h50的初步毒性,发现与模型组相比,给药组的体重无统计学差异,he染色发现两组间重要器官的显微结构亦无显著差异(详情见实施例11)。
本发明所述小分子化合物l16h50可以应用于制备与fgfr1高表达相关肿瘤的抗肿瘤药物及与肿瘤相关疾病的治疗药物中,所述肿瘤疾病如下:乳腺癌、肾癌、膀胱癌、口腔癌、咽喉癌、胃癌、结肠癌、宫颈癌、卵巢癌、皮肤癌、肝癌、多发性骨髓瘤、前列腺癌、脑肿瘤。
本发明还提供了一种用于治疗肿瘤的药物组合物,其含有治疗有效量的作为活性成分的以上所述小分子化合物或其可药用盐及其药用辅料。“药物组合物”指本发明所述小分子化合物或其可药用盐与现已上市的抗肿瘤药物联合使用,制备得到的防治肿瘤类药物的组合物,已上市的抗肿瘤药物包括各种细胞毒性药物和靶向药物。
本文中所用“药用辅料”指药学领域常规的药物载体,例如:稀释剂、赋形剂如水等,填充剂如淀粉、蔗糖等;粘合剂如纤维素衍生物、藻酸盐、明胶和聚乙烯吡咯烷酮;湿润剂如甘油;崩解剂如琼脂、碳酸钙和碳酸氢钠;吸收促进剂如季铵化合物;表面活性剂如十六烷醇;吸附载体如高岭土和皂粘土;润滑剂如滑石粉、硬脂酸钙/镁、聚乙二醇等。另外还可以在组合物中加入其它辅剂如香味剂、甜味剂等。
本发明药物组合物的各种剂型可以按照药学领域的常规生产方法制备。例如使活性成分与一种或多种载体混合,然后将其制成所需的剂型。所述药物的制剂形式包括注射剂、片剂、胶囊剂、气雾剂、栓剂、膜剂、滴丸剂、软膏剂、控释或缓释剂或纳米制剂。本发明可以组合物的形式通过口服,鼻吸入、直肠或者肠胃外给药的方式施用于需要这种治疗的患者。用于口服时,可将其制成常规的固体制剂如片剂、粉剂、粒剂、胶囊等,制成液体制剂如水或油悬浮剂或其它液体制剂如糖浆等;用于肠胃外给药时,可将其制成注射用的溶液、水或油性悬浮剂等。
附图说明
图1小分子化合物l16h50非atp竞争性地抑制fgfr1的激活。a.l16h50的结构及其对fgfr1激酶抑制活性的半数抑制浓度(ic50)。b.将fgfr1高表达的hek-293t细胞(只表达fgfr1)铺板,用无血清的dmem高糖培养基饥饿12小时后,更新培养液并加入各种浓度的受测化合物预处理1小时后,再加入40ng/mlfgf2刺激10分钟。裂解蛋白,westernblot法检测fgfr1的磷酸化水平。c.将sgc7901细胞铺板,用无血清的rpmi-1640培养基饥饿12小时后,更新培养液并加入各种浓度的受测化合物预处理1小时后,再加入40ng/mlfgf2刺激10分钟。裂解蛋白,westernblot法检测fgfr1及其下游蛋白包括frs2α、akt、erk1/2及plcγ的磷酸化水平。d.l16h50的atp竞争性实验。
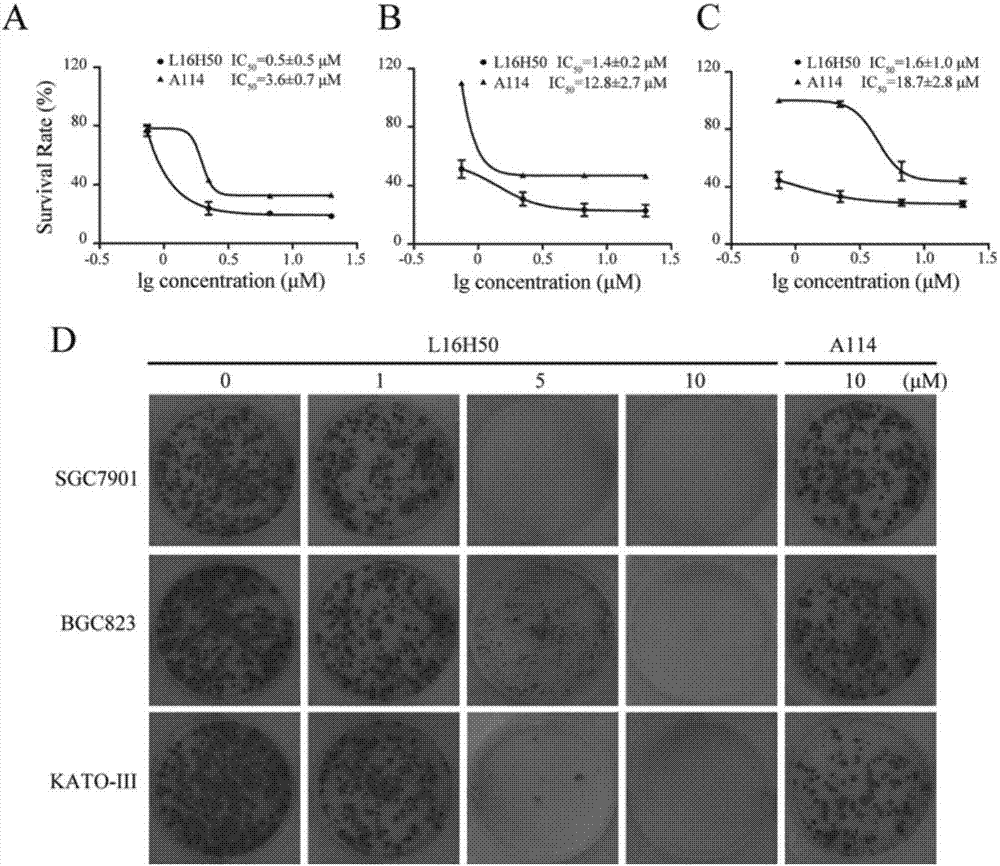
图2l16h50抑制胃癌细胞的生长。(a-c)细胞用不同浓度的化合物处理72小时,再用mtt法检测细胞生存率。d.用集落形成实验检测l16h50对胃癌细胞集落形成能力的影响。
图3l16h50将胃癌细胞周期阻滞在g2/m期。a和b.流式细胞术检测不同浓度(1,5,10μm)的l16h50作用10小时后胃癌细胞周期分布的变化。c.westernblot法检测不同浓度(1,5,10μm)的l16h50作用10小时后胃癌细胞中周期相关蛋白cyclinb1和cdc2p34水平的改变。三次重复实验得到相同结论(*p<0.05,**p<0.01,***p<0.001,vs0group)。
图4l16h50诱导胃癌细胞凋亡。a-d流式细胞术检测不同浓度(1,5,10μm)的l16h50和10μm的a114对胃癌细胞作用12小时后,对胃癌细胞凋亡情况的影响。e.caspase-3/caspase-9检测试剂盒检测10μm的l16h50和a114作用12小时后对胃癌细胞中caspase-3/caspase-9活性的影响。f-hwesternblot法不同浓度(1,5,10μm)的l16h50和10μm的a114作用12小时后胃癌细胞中bcl-2和cleaved-parp水平的变化。每个柱条为三次实验的平均值和误差值(*p<0.05,**p<0.01,***p<0.001,vsdmsogroup)。
图5l16h50的裸鼠体内抗胃癌活性。a-d将7x106的sgc7901细胞接种于裸鼠右肩区域以建立异位瘤模型,接种7天后开始腹腔注射给药(10mg/kg受试药:l16h50、azd4547),开始给药后每两天测肿瘤长短径及鼠重,给药20天后处死小鼠,剥出肿瘤称瘤重。e和f.westernblot法检测肿瘤组织中fgfr1及其下游蛋白包括frs2α、akt及erk1/2的磷酸化水平。g.ihc检测肿瘤组织中ki67和bcl-2的水平(*p<0.05,**p<0.01,***p<0.001,vsvehiclegroup)。
图6l16h50对icr小鼠的初步急性毒性作用。icr小鼠用vehicle(saline)或1g/kg的l16h50口服给药后,连续14天每隔1天记录体重(b)。14天后处死小鼠,取出重要脏器称重(a)并用he染色了解脏器显微结构的变化(c)。
具体实施方式
本发明在以下的实施例中进一步说明。这些实施例只是为了说明本发明的目的,而不是用来限制本发明的范围。
实施例1化合物的合成
将1mmol相应的对苯二甲醛和2mmol取代苯乙酮溶于20ml左右的无水乙醇中,5-8℃下加入50%naoh溶液10-15滴,5-8℃反应5-24h后,用tlc检测反应的进行。反应完后,加入1-2倍反应液体积的水,析出沉淀,过滤,真空干燥过夜后得粉末状产物,经硅胶柱色谱纯化后得纯度均大于98%的化合物。在合成的多个化合物中,药理活性差别很大,有代表性的化合物及其理化性质如下所述:
有效化合物(2e,2'e)-3,3'-(1,4-phenylene)bis(1-(2-chlorophenyl)prop-2-en-1-one)(l16h50)yellowpowder,90.2%yield,mp144.4~147.1℃.esi-msm/z:407.0(m+1)+calcdforc24h16cl2o2:407.29.1h-nmr(dmso),δ:7.601(s,4h,h-2,h-3,h-4,h-6),7.426~7.515(m,8h,h-3’×2,h-4’×2,h-6’×2,h-β×2),7.376(t,j=7.2hz,2h,h-5’×2),7.186(d,j=15.6hz,2h,h-α×2).
无效化合物(2e,2'e)-3,3'-(1,4-phenylene)bis(1-(2-fluorophenyl)prop-2-en-1-one)(l15h50)earthyellowpowder,91.3%yield,mp133.8~135.1℃.esi-msm/z:374.9(m+1)+calcdforc24h16f2o2:374.38.1h-nmr(dmso),δ:7.874(s,4h,h-2,h-3,h-5,h-6),7.814(m,j=1.8hz,2h,h-4’,h-4’),7.724(dd,j=15.6hz,2h,h-β×2),7.666(m,2h,h-5’,h-5’),7.545(dd,j=15.2hz,2h,h-α×2),7.371(t,j=7.2hz,2h,h-3’,h-3’),7.312(m,j=2.4hz,8.4hz,2h,h-2’,h-2’).
无效化合物(2e,2'e)-3,3'-(1,4-phenylene)bis(1-(4-hydroxy-3-methoxyphenyl)prop-2-en-1-one)(l20h50)brownpowder,70.2%yield,mp91.6~93.2℃.esi-msm/z:428.9(m-1)-calcdforc26h22o6:430.45.1h-nmr(dmso),δ:7.703(s,2h,h-6’×2),7.477(dd,j=15.6hz,2h,h-α×2),7.451(dd,j=15.6hz,2h,h-β×2),7.382(t,4h,h-2’×2,h-3’×2),7.260(s,3h,h-5’×2,h-2),7.163(d,3h,h-4,h-5,h-6),3.814(s,6h).
实施例2化合物对fgfr1激酶的抑制活性
采用caliperezreader药物筛选平台检测化合物对fgfr1的激酶活性。将atp的浓度设置在fgfr1的km值,即262μmol/ml。为了检测化合物对激酶抑制活性的ic50,将化合物稀释成从5nmol/ml到100μmol/ml的10个浓度梯度。在迁移率检测时,产物的积累量用转化率=a/(a+b)表示,a代表产物峰高度,b代表底物峰高度。l16h50对激酶抑制活性的ic50为:3.8±2.7μm;而对照化合物l15h50和l20h50在100μmol/ml浓度下,对fgfr1激酶的抑制率分别为19.5%、15.8%,可以认为两者均不是fgfr1激酶抑制剂。
实施例3l16h50以非atp竞争性方式抑制fgfr1激酶活性
l16h50为一种全新型的fgfr1激酶抑制剂,为了探究其对fgfr1激酶的抑制模式,测定了其与atp的竞争关系。具体方法:首先固定底物的浓度(相对激酶来说是足量的浓度),然后加入一定量的激酶,再分别加入不同浓度的atp(5000、2500、1250、625、312.5、156.25、78.125和39.0625μmol/ml)和化合物(l16h50:100、50、25、13、6.5、3.3、1.6、0.8、0.4和0.2μmol/ml),然后测定不同浓度的atp和化合物条件下,激酶对底物的转化率,根据公式转化率=(vmax*x)/{km*[(1+i/ki)n]+x}。x代表atp的浓度,n代表希尔系数。实验结果见图1d,结果显示,在固定l16h50浓度下,随着atp浓度的增加,底物转化率未表现出增加的趋势,因此atp的浓度的变化并未对激酶的底物转化率表现出明显影响,说明其为非atp竞争性抑制剂。
实施例4westernblot法测定l16h50对细胞中fgfr1信号通路的抑制作用
为了研究化合物对细胞中fgfr1激酶的抑制作用,通过westernblot法测定了化合物对人工构建高表达fgfr1的hek-293t、及内源高表达fgfr1的sgc7901细胞中fgfr1及其下游信号通路蛋白的影响。具体方法:取对数期生长的sgc7901或者hek-293tfgfr1细胞(只表达fgfr1)按每孔30万个细胞铺在6孔板中,贴壁过夜后,再用无血清的培养基饥饿12小时,再分(空白组、fgf240ng/ml组、fgf2+l16h50/a114/azd4547组)组加药。具体加药方法:先加入化合物预孵1小时后,再加入40ng/mlfgf2刺激10分钟,收蛋白,用westernblot测定fgfr1信号通路蛋白的表达情况。结果见图1b和1c。l16h50对hek-293t中fgf2诱导的fgfr1的磷酸化表现出浓度剂量依赖性的抑制作用;对sgc7901中fgf2诱导的fgfr1,及其下游信号通路蛋白frs2α、plcγ、erk、akt的磷酸化均表现出很好的抑制作用,且呈现出浓度剂量依赖性。在浓度10μm浓度下,l16h50在以上活性中均表现出与阳性药物azd4547和a114相当的活性。
实施例6mtt法测定化合物对细胞生长的影响
fgfr1与肿瘤细胞的生长密切相关,因此通过mtt法测定了化合物对肿瘤细胞的生长抑制活性。具体方法:将处于对数期生长的胃癌细胞sgc7901、bgc823及kato-iii细胞消化并按3000个/孔各自铺在96孔板中,贴壁过夜,换液,按(空白组、dmso组、l16h50/a114组)组加药。作用72小时后,观察细胞存活情况并在每个孔加入25μlmtt(浓度为5mg/ml),置于37℃、5%co2培养箱培养4-6小时,吸取上清,留下结晶,每孔加入150μldmso溶解结晶,室温避光条件下在微量振荡仪上振荡10min,观察结晶溶解情况,待结晶完全溶解后在酶标仪490nm波长处读取od值,并计算抑制率和ic50。结果见图2a-c。在sgc7901,bgc823及kato-iii三株细胞株中,l16h50浓度依赖且时间依赖地抑制胃癌细胞的增殖,其抑制效果明显优于对照a114。l16h50对sgc7901、bgc823及kato-iii的半数抑制浓度(ic50)分别为0.5±0.4,1.4±0.2和1.6±1.0μm。
实施例7集落形成实验
用集落克隆实验测定了化合物对胃癌细胞集落形成的影响。具体方法:sgc7901、bgc803及kato-iii细胞按300个/孔接种至6孔板,贴壁过夜,换液,分(空白组、l16h50组)组加药作用8小时后,换成新鲜的完全培养基,在37℃、5%co2的环境中培养3周。将预处理的细胞从恒温培养箱中取出,弃培养基,1mlpbs洗涤细胞2次,用4%多聚甲醛室温下固定10分钟后,吸去多聚甲醛,pbs洗去残余多聚甲醛,加入0.5ml结晶紫染色液室温下染30分钟,回收结晶紫染色液(室温保存,可重复利用)。pbs洗去残留的结晶紫染色液,晾干后用单反照相机记录图片。实验结果见图2d。结果显示l16h50作用8小时后,对三种胃癌细胞均能以浓度剂量依赖性地抑制肿瘤细胞集落的形成,在浓度5μm下就表现出良好的抑制作用,10μm的l16h50处理过的胃癌细胞基本上不能形成集落,在相同浓度下其活性明显由于对照a114。
实施例8对肿瘤细胞周期的阻滞
用流式细胞仪测定了l16h50对胃癌细胞周期的影响。具体方法,:sgc7901、bgc803及kato-iii细胞按30万个/孔接种至6孔板,贴壁过夜后,换液,分(空白组、l16h50组)组加药作用10小时。从培养箱中取出预处理过的细胞,吸去培养基,每孔加入1mlpbs洗涤细胞,吸去pbs,重复1次,加入含edta的胰蛋白酶消化细胞,注意吹打轻柔,收集细胞至5mlep管中,离心(1000转、5分钟),弃上清,加入1mlpbs后轻轻吹打重悬,再次离心。离心后固定,先加入1mlpbs重悬,再在振荡器上边振荡边缓缓加入3ml预冷的乙醇。混匀后置于-20℃环境下4小时以上,固定完毕后,离心以去掉乙醇,再加入1mlpbs重悬离心,洗去残留的乙醇。离心后,去上清,加入0.5mlpi(含rnase)在4℃避光的环境下孵育10分钟后,再用200目的纱布将染色的细胞过滤至流式上样管,按流式周期操作步骤上机检测。结果见图3a-b。三种胃癌细胞中加入1μm的l16h50后,g2/m期细胞的比例均明显上升,在10μm时g2/m期细胞比例均提高到大于20%的水平。因此l16h50能够通过阻滞细胞周期在g2/m期,起到抑制肿瘤细胞生长的作用。
cyclinb1和cdc2p34这两个蛋白主要调控g2/m期,因此用westernblot测定了其对这两种周期蛋白的影响。结果见图3c,l16h50能够剂量依赖地下调cyclinb1和cdc2p34的水平,在10μm时对两种蛋白的表达几乎被完全抑制。
实施例9诱导肿瘤细胞凋亡
为了进一步了解l16h50对胃癌细胞的作用,我们检测了l16h50对细胞凋亡的作用。结果见图4。流式细胞仪测定凋亡的方法:sgc7901、bgc803及kato-iii细胞按50万个/孔接种至6孔板,贴壁过夜后,换液,分(空白组、l16h50/a114组)组加药作用12小时。从培养箱中取出预处理过的细胞,收集培养基至流式上样管,每孔加入1mlpbs洗涤细胞,吸取pbs至对应流式上样管,重复1次,加入不含edta的胰蛋白酶消化细胞,注意吹打轻柔,收集细胞至对应流式上样管中,离心(1000转,5分钟),弃上清,加入1mlpbs后轻轻吹打重悬,再次离心。离心后去上清,将结合缓冲液(10×bindingbuffer)用pbs配置成1×bindingbuffer,除低浓度药物组外每组加入0.1ml1×bindingbuffer重悬,低浓度药物组则加入0.4ml1×bindingbuffer重悬并分成4管(每管0.1ml细胞悬液,分别标记为未染色管、低浓度双染管、annexin-v管及pi管),除低浓度药物组外每组加入3μl的annexin-v避光染色10分钟,同时取低浓度双染管及annexin-v管加入3μl的annexin-v避光染色10分钟,接着再在除低浓度药物组外每组加入1μl的pi避光染色5分钟,同时取低浓度双染管及pi管加入1μl的pi避光染色5分钟,染色完成后,每管加入400μl1×bindingbuffer混匀,按流式凋亡操作步骤上机检测。流式细胞仪测定结果显示了l16h50显著地上调了三株胃癌细胞(包括sgc7901,bgc823及kato-iii)的凋亡水平(图4a-d)。10μm的l16h50引起的凋亡比例在sgc7901、bgc823及kato-iii分别占到24.1%、16.81%和12.2%。
此外,检测了化合物对凋亡蛋白caspase3/9活力的影响。方法:将处于对数期的sgc7901按100万个/孔接种至6孔板,贴壁过夜后,换液,分(空白组、10μml16h50/10μma114组)组加药作用12小时。caspase3/9的活性检测步骤按试剂盒说明书操作。结果显示l16h50能够明显地增强caspase3/9的活性(图4e)。进一步用westernblot对凋亡相关蛋白进行了检测(图4f-h)。发现在这三株胃癌细胞株中l16h50显著地上调抗凋亡蛋白bcl-2的水平,同时上调了凋亡蛋白cleaved-parp的水平。
实施例10裸鼠异位荷瘤实验
上述结果表明了l16h50确实具有较好的体外抗肿瘤效果,因此用sgc7901的异位瘤模型进一步评估l16h50在裸鼠体内抗肿瘤的效果。方法:将处于对数期的sgc7901细胞用pbs(按700万个细胞/0.1mlpbs)重悬混匀,用1ml注射器将0.1ml细胞悬液皮下注射至nu/nubalb/c裸鼠的右肩区域以建立异位瘤模型,根据裸鼠体重将裸鼠随机分为模型组、给药组,每组7只。药物用剂型(pbs:聚乙二醇:蓖麻油=7:2:1)溶解,1周后,模型组每天腹腔注射剂型0.1ml,药物组每天腹腔注射药物10mg/kg(受试药:l16h50、azd4547)。给药期间每2天测定裸鼠体重、肿瘤长径(l)及短径(w),肿瘤体积计算公式:肿瘤体积(v)=0.5×w2×l。给药20天后,颈椎脱臼法处死裸鼠,剥出肿瘤并称重,之后将肿瘤分成两部分:一部分-80℃保存,一部分福尔马林固定。结果见图5。l16h50显著地减少了肿瘤的体积和重量,并且这效果不弱于azd4547。为了检测l16h50在体内对fgfr1的作用,我们用westernblot检测了肿瘤组织中的fgfr1及相关下游蛋白。westernblot结果显示l16h50显著地抑制了fgfr1、frs2、akt及erk的激活。此外,我们还用免疫组化的方法对肿瘤组织中的增殖及凋亡相关蛋白进行测定。结果显示,l16h50明显下调了增殖相关因子ki67和抗凋亡相关因子bcl-2的水平。
实施例11体内毒性实验
在异位瘤模型给药期间,每2天检测裸鼠的体重,发现体重并没有因为给药发生显著的改变(图5d)。此外进行了化合物对的初步的急性毒性研究。方法:取20g左右的icr小鼠分为模型组、给药组,每组7只,一次性灌胃给予药物(受试药:l16h50)1g/kg。给药后每天测量小鼠体重,连续观察14天后,颈椎脱臼法处死小鼠,取出心肝脾肺肾胃,观察脏器有无异常并称重,之后脏器用福尔马林固定。结果显示(图6),1g/kg的剂量给药并没有对icr小鼠的体重造成明显变化(图6b),并且造模组和给药组之间的内脏重量(图6a),及he染色显示的显微结构(图6c)均无明显差异。由以上实验说明在给药剂量范围内,l16h50并未表现出明显的小鼠体内毒性。
- 还没有人留言评论。精彩留言会获得点赞!