一种偕二氟取代吡咯烷酮化合物的合成方法与流程
技术领域:
本发明属于化学有机合成技术领域,具体涉及一种以烯烃和溴代二氟乙酰胺类化合物为原料,催化合成偕二氟取代吡咯烷酮化合物的方法。
背景技术:
:
含氮杂环化合物广泛应用于医药、农用化学品和材料领域,引入氟原子或含氟分子可以提高其亲脂性和代谢稳定性,改善生物医药的药效((a)uneyama,k.;sasaki,k.pharmaceuticalscontainingfluorinatedheterocycliccompounds.influorinatedheterocycliccompounds:synthesis,chemistryandapplication;petrov,v.a.,ed.;johnwiley&sons:hoboken,2009;419.(b)nosova,e.v.;lipunova,g.n.;charushin,v.n.;chupakhin,o.n.j.fluorinechem.2010,131,1267.)。其中,以吡咯烷酮为母核结构的化合物因其具有消炎、抗老年痴呆、抗肿瘤及抗hiv等药理活性,在医学和药物研究中引起了极大的兴趣((a)simonshorvon.thelancet,2001,358,1885.(b)farina,c.;gagliardi,s.;ghelardini,c.;martinelli,m.;norcini,.m.;parini,c.;petrillo,p.;ronzoni,s.bioorganic&medicinalchemistry,2008,16,3224.)。在吡咯烷酮分子中引入氟原子将进一步改善药品的生理活性,并且含有偕二氟取代吡咯烷酮衍生物也已被证明具有抗消炎、抗肿瘤、抗癌的作用。因此,发展绿色环保、新型高效的合成偕二氟取代吡咯烷酮化合物的方法是相当重要且具有重要药用研究价值。
此前,合成偕二氟取代吡咯烷酮化合物的方法鲜有报道。经过文献检索,目前合成该类化合物的方法,步骤较为复杂,且总转化效率较低,同时该方法的适用范围不够宽泛((a)nguyen,q.p.b.;kim,b.m.;song,m.s.;yoon,k.h.;chai,k.y.bull.koreanchem.soc.2014,35,313.)。近年来,烯烃的双官能团化反应由于可以同时在分子中引入两个不同的基团,所用原料简单易得,原子经济性高,在合成复杂的有机分子中得到了迅速的发展。因此,发展高效、廉价、环保的利用烯烃的双官能团化反应来合成偕二氟取代吡咯烷酮化合物极其重要。
到目前为止,通过烯烃与溴代二氟乙酰胺类化合物反应,由廉价易得的铜盐作为催化剂来合成偕二氟取代吡咯烷酮化合物的方法还未见文献报道。
技术实现要素:
:
本发明目的在于提供一种以烯烃和溴代二氟乙酰胺类化合物为原料,催化合成偕二氟取代吡咯烷酮化合物的方法,该方法具有原子经济性高、反应步骤简单、原料廉价易得、底物适用范围广泛等特点。
一种偕二氟取代吡咯烷酮化合物的合成方法,其包括以下步骤:
将溴代二氟乙酰胺类化合物用溶剂溶解,并加入烯烃混合均匀,然后加入配体邻菲罗啉(phen)、催化剂cui和碱k2co3,在密闭条件下升温至110±10℃搅拌反应1-3小时,反应结束经后处理即得。反应方程式如下所示:
式中,化合物1为烯烃,可以是苯乙烯类化合物、烯丙基类化合物、烯基醚类化合物、n-烯基杂环化合物以及非活性烯烃类化合物等,具体的如苯乙烯类化合物(如苯乙烯、4-硝基苯乙烯)、丁香酚甲醚或环己基乙烯基醚类化合物、n-乙烯基杂环化合物等。化合物2为溴代二氟乙酰胺类化合物,可以是芳环或萘环的溴代二氟乙酰胺类化合物等,具体的如溴代二氟乙酰对甲苯胺、溴代二氟乙酰苯胺或溴代二氟乙酰-2-萘胺等二氟乙酰胺类化合物。所用溶剂可以是乙腈(ch3cn)等,1mmol溴代二氟乙酰胺类化合物添加8-15ml溶剂为宜。
具体的,所述溴代二氟乙酰胺类化合物与烯烃的摩尔比为1:1.1-1:1.5。
具体的,所述配体邻菲罗啉(phen)与溴代二氟乙酰胺类化合物的摩尔比为5—15:100,催化剂cui与溴代二氟乙酰胺类化合物的摩尔比为5—15:100,碱k2co3与溴代二氟乙酰胺类化合物的摩尔比为1.5—2.5:1。
本发明以cui为催化剂、邻菲罗啉(phen)为配体,k2co3为碱在乙腈溶剂中进行反应,由烯烃和溴代二氟乙酰胺类化合物经催化环加成一步得到偕二氟取代吡咯烷酮化合物。和现有技术相比,本发明的优点和创新性在于:1)克服了目前仅有的通过多步反应合成偕二氟取代吡咯烷酮化合物方法存在步骤繁琐的缺陷,本发明方法反应一步合成,操作简单,产率较高。2)该反应所用的原料易从工业产品中获得,试剂廉价,底物范围宽泛,适用于工业生产。
附图说明
图1为实施例1产物3,3-二氟-4-吡咯啉-2-酮3a的1hnmr谱图;
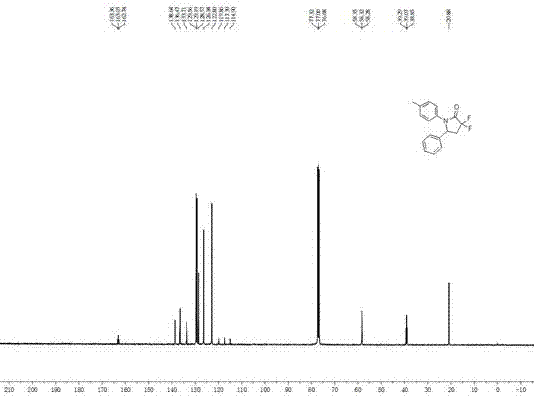
图2为实施例1产物3,3-二氟-4-吡咯啉-2-酮3a的13cnmr谱图;
图3为实施例1产物3,3-二氟-4-吡咯啉-2-酮3a的19fnmr谱图;
图4为实施例4产物3,3-二氟-4-吡咯啉-2-酮3d的1hnmr谱图;
图5为实施例4产物3,3-二氟-4-吡咯啉-2-酮3d的13cnmr谱图;
图6为实施例4产物3,3-二氟-4-吡咯啉-2-酮3d的19fnmr谱图。
具体实施方式
以下结合实施例对本发明的技术方案作进一步地详细介绍,但本发明的保护范围并不局限于此。
实施例1:
向装有搅拌子的35ml密封管中,依次加入79.2mg(0.3mmol)溴代二氟乙酰对甲苯胺,3.0ml乙腈,41μl苯乙烯(0.36mmol),混合均匀。然后加入5.4mgphen(0.03mmol)、5.7mgcui(0.03mmol)和82.9mgk2co3(0.6mmol)。将管口密封,加热至110℃搅拌反应2小时(薄层色谱tlc监测反应进程)。反应结束后,冷却到室温,向反应混合物中加入3ml蒸馏水,乙酸乙酯萃取(5ml×3),合并有机相,无水硫酸钠干燥,减压蒸馏除去溶剂,剩余物经硅胶柱层析(v石油醚:v乙酸乙酯=30:1)分离得74.8mg白色固体产物3a,产率87%。反应见下式:
谱图解析数据
whitesolid.mp:139-140ºc.1hnmr(400mhz,cdcl3):δ=2.26(s,3h),2.48-2.60(m,1h),3.05-3.18(m,1h),5.26-5.30(m,1h),7.08(d,j=8.4hz,2h),7.23(d,j=8.8hz,2h),7.28-7.34(m,5h);13cnmr(100mhz,cdcl3):δ=20.9,39.1(t,j=22.0hz),58.3(t,j=4.0hz),117.4(t,j=248.0hz),122.8,126.3,128.6,129.2,129.6,133.7,136.5,138.6,163.1(t,j=31.0hz);19fnmr(376mhz,cdcl3):δ=-104.4(d,j=270.0hz,1f),-101.2(d,j=270.7hz,1f).hrms(esi-tof)calcdforc17h16f2no,[m+h]+m/z288.1200,found288.1208。
实施例2:
向装有搅拌子的35ml密封管中,依次加入79.2mg(0.3mmol)溴代二氟乙酰对甲苯胺,3.0ml乙腈,53.6mg4-硝基苯乙烯(0.36mmol),混合均匀。然后加入5.4mgphen(0.03mmol)、5.7mgcui(0.03mmol)和82.9mgk2co3(0.6mmol)。将管口密封,加热至110℃搅拌反应2小时(薄层色谱tlc监测反应进程)。反应结束后,冷却到室温,向反应混合物中加入3ml蒸馏水,乙酸乙酯萃取(5ml×3),合并有机相,无水硫酸钠干燥,减压蒸馏除去溶剂,剩余物经硅胶柱层析(v石油醚:v乙酸乙酯=30:1)分离得91.7mg白色固体产物3b,产率92%。反应见下式:
谱图解析数据
whitesolid.mp:144-146ºc.1hnmr(400mhz,cdcl3):δ=2.26(s,3h),2.46-2.58(m,1h),3.12-3.25(m,1h),5.46-5.50(m,1h),7.10(d,j=8.4hz,2h),7.28(d,j=8.4hz,2h),7.43(d,j=8.4hz,2h),8.16(d,j=8.4hz,2h);13cnmr(100mhz,cdcl3):δ=20.8,38.4(t,j=22.0hz),57.4,116.9(t,j=248.0hz),122.4,124.4,127.4,129.8,133.1,137.0,145.8,147.9,162.7(t,j=31.0hz);19fnmr(376mhz,cdcl3):δ=-104.8(d,j=270.7hz,1f),-100.4(d,j=270.7hz,1f).hrms(esi-tof)calcdforc17h15f2n2o3,[m+h]+m/z333.1051,found333.1059。
实施例3:
向装有搅拌子的35ml密封管中,依次加入79.2mg(0.3mmol)溴代二氟乙酰对甲苯胺,3.0ml乙腈,62μl丁香酚甲醚(0.36mmol),混合均匀。然后加入5.4mgphen(0.03mmol)、5.7mgcui(0.03mmol)和82.9mgk2co3(0.6mmol)。将管口密封,加热至110℃搅拌反应2小时(薄层色谱tlc监测反应进程)。反应结束后,冷却到室温,向反应混合物中加入3ml蒸馏水,乙酸乙酯萃取(5ml×3),合并有机相,无水硫酸钠干燥,减压蒸馏除去溶剂,剩余物经硅胶柱层析(v石油醚:v乙酸乙酯=30:1)分离得99.7mg白色固体产物3c,产率60%。反应见下式:
谱图解析数据
whitesolid.mp:135-136ºc.1hnmr(400mhz,cdcl3):δ=2.34-2.62(m,6h),3.05(dd,j1=2.4hz,j2=14.0hz,1h),3.85(s,3h),3.86(s,3h),4.40-4.45(m,1h),6.55(s,1h),6.66(d,j=8.0hz,1h),6.80(d,j=8.4hz,1h),7.28(d,j=8.0hz,2h),7.39(d,j=8.4hz,2h);13cnmr(100mhz,cdcl3):δ=21.0,33.8(t,j=22.0hz),38.8,55.6(d,j=23.0hz),55.8,55.9,111.5,112.3,115.1(t,j=248.0hz),121.3,123.6,127.8,130.0,133.1,137.2,148.2,149.1,162.6(t,j=32.0hz);19fnmr(376mhz,cdcl3):δ=-104.9(d,j=270.0hz,1f),-100.7(d,j=270.7hz,1f).hrms(esi-tof)calcdforc20h22f2no3,[m+h]+m/z362.1568,found362.1573。
实施例4:
向装有搅拌子的35ml密封管中,依次加入79.2mg(0.3mmol)溴代二氟乙酰对甲苯胺,3.0ml乙腈,85μl环己基乙烯基醚(0.6mmol),混合均匀。然后加入5.4mgphen(0.03mmol)、5.7mgcui(0.03mmol)和82.9mgk2co3(0.6mmol)。将管口密封,加热至110℃搅拌反应2小时(薄层色谱tlc监测反应进程)。反应结束后,冷却到室温,向反应混合物中加入3ml蒸馏水,乙酸乙酯萃取(5ml×3),合并有机相,无水硫酸钠干燥,减压蒸馏除去溶剂,剩余物经硅胶柱层析(v石油醚:v乙酸乙酯=40:1)分离得87.2mg白色固体产物3d,产率94%。反应见下式:
谱图解析数据
whitesolid.mp:108-109ºc.1hnmr(400mhz,cdcl3):δ=1.09-1.47(m,6h),1.60-1.76(m,4h),2.36(s,3h),2.50-2.60(m,1h),2.76-2.88(m,1h),3.25-3.31(m,1h),5.41-5.43(m,1h),7.22(d,j=8.0hz,2h),7.32(d,j=8.4hz,2h);13cnmr(100mhz,cdcl3):δ=21.1,23.7(d,j=3.0hz),25.3,32.1,32.8,38.5(t,j=22.0hz),84.4(d,j=5.0hz),116.7(t,j=248.0hz),124.6,129.8,133.3,137.7,162.7(t,j=31.0hz),175.8;19fnmr(376mhz,cdcl3):δ=-105.5(d,j=274.5hz,1f),-100.9(d,j=270.7hz,1f).hrms(esi-tof)calcdforc17h22f2no2,[m+h]+m/z310.1619,found310.1616。
实施例5:
向装有搅拌子的35ml密封管中,依次加入75.0mg(0.3mmol)溴代二氟乙酰苯胺,3.0ml乙腈,41μl苯乙烯(0.36mmol),混合均匀。然后加入5.4mgphen(0.03mmol)、5.7mgcui(0.03mmol)和82.9mgk2co3(0.6mmol)。将密封管口封闭,加热至110℃搅拌反应2小时(薄层色谱tlc监测反应进程)。反应结束后,冷却到室温,向反应混合物中加入3ml蒸馏水,乙酸乙酯萃取(5ml×3),合并有机相,无水硫酸钠干燥,减压蒸馏除去溶剂,剩余物经硅胶柱层析(v石油醚:v乙酸乙酯=30:1)分离得69.7mg白色固体产物3e,产率85%。反应见下式:
谱图解析数据
whitesolid.mp:125-127ºc.1hnmr(400mhz,cdcl3):δ=2.48-2.60(m,1h),3.05-3.18(m,1h),5.29-5.33(m,1h),7.08-7.16(m,2h),7.21-7.24(m,3h),7.28-7.33(m,3h),7.41(d,j=8.4hz,2h);13cnmr(100mhz,cdcl3):δ=39.1(t,j=22.0hz),58.2(d,j=4.0hz),117.3(t,j=248.0hz),122.8,126.3,126.6,128.6,129.0,129.2,136.3,138.5,163.1(t,j=31.0hz);19fnmr(376mhz,cdcl3):δ=-104.4(d,j=270.7hz,1f),-101.5(d,j=270.7hz,1f).hrms(esi-tof)calcdforc16h14f2no,[m+h]+m/z274.1043,found274.1044。
实施例6:
向装有搅拌子的35ml密封管中,依次加入90.0mg(0.3mmol)溴代二氟乙酰-2-萘胺,3.0ml乙腈,41μl苯乙烯(0.36mmol),混合均匀,然后加入5.4mgphen(0.03mmol)、5.7mgcui(0.03mmol)和82.9mgk2co3(0.6mmol)。将管口密封,加热至110℃搅拌反应2小时(薄层色谱tlc监测反应进程)。反应结束后,冷却到室温,向反应混合物中加入3ml蒸馏水,乙酸乙酯萃取(5ml×3),合并有机相,无水硫酸钠干燥,减压蒸馏除去溶剂,剩余物经硅胶柱层析(v石油醚:v乙酸乙酯=30:1)分离得75.6mg白色固体产物3f,产率78%。反应见下式:
谱图解析数据
whitesolid.mp:162-164ºc.1hnmr(400mhz,cdcl3):δ=2.54-2.66(m,1h),3.11-3.24(m,1h),5.44-5.48(m,1h),7.23-7.32(m,5h),7.42-7.47(m,2h),7.56(dd,j1=2.0hz,j2=9.2hz,1h),7.74-7.77(m,3h),7.91(d,j=1.6hz,1h);13cnmr(100mhz,cdcl3):δ=39.1(t,j=22.0hz),58.4,117.4(t,j=248.0hz),121.0,121.4,126.2,126.3,126.6,127.5,127.9,128.6,128.9,129.2,131.5,133.1,133.7,138.4,163.3(t,j=31.0hz);19fnmr(376mhz,cdcl3):δ=-104.3(d,j=270.7hz,1f),-101.2(d,j=270.7hz,1f).hrms(esi-tof)calcdforc20h16f2no,[m+h]+m/z324.1200,found324.1196。
- 还没有人留言评论。精彩留言会获得点赞!