与多重基因组获得和丢失分析相关的方法和组合物与流程
本申请为分案申请,其原申请的申请日为2008年11月24日,申请号为200880126212.7,名称为“与多重基因组获得和丢失分析相关的方法和组合物”。
相关申请的参引
本申请要求2008年11月21日提交的流水号为12/275,895的美国专利申请的优先权,所述美国专利申请为2008年3月26日提交的流水号为12/055,919的美国专利申请的部分继续申请,美国专利申请no.12/055,919要求2007年12月5日提交的流水号为60/992,489的美国临时专利申请的优先权。每一份申请的全部内容通过参引纳入本申请。
本文描述的技术大体涉及核酸检测用方法和组合物。更具体而言,所描述的是基因组获得和丢失分析用方法和组合物。
背景技术:
通过对基因组获得和丢失的检测分析可以对构成疾病、行为和认知病症以及其他遗传病变的病因的遗传性异常进行检测和诊断。
array-cgh(acgh)是一种多重分析方法,其中固定的dna探针捕获来自测试样本和参照样本的经标记的互补基因组dna序列。每一个探针都会产生一个由探针序列界定的组成序列中的一个序列的测试量/参照量的比值。在acgh中,通常使用细菌人工染色体(bac)作为固定到阵列固体支持物上的探针材料。典型的bac序列长度介于大约100kb至大约250kb之间,通常的长度为175kb。这一长度的探针非常适合acgh,原因在于它们长到足以形成大的杂交信号,并且不会对小的突变如snp产生响应,然而又短到足以提供充分的基因组解析力,从而较为精确地检测基因组获得或者丢失的断点(breakpoint)。例如,一个设计精良的bac阵列可以检测微缺失综合征如1p36缺失综合征或digeorge综合征中不同大小的缺失区域,其中跨越疾病相关区域的不同部分的缺失可以对应不同的表型。
所谓组成型acgh微阵列的设计正趋于使用多种单独的探针,例如与先天dna障碍相关的每一个基因组区域中的bac探针,从而以较高的解析力检测基因组获得和丢失的程度。较高的解析力会揭示出更多有关获得或丢失区域的确切大小和边界的信息。但是,存在可能并不需要对获得或者丢失区域的确切大小和边界进行高解析力检测的情况。
非整倍性是基因组获得-丢失异常的实例,其中以高解析力检测断点不再可行。非整倍性是一条完整的染色体(平均大小超过100mb)的获得(最为典型,如三体性)或丢失,这与微缺失障碍相反,在后者中,缺失区域的典型长度可能仅1至10mb左右。在acgh分析中,非整倍性检测(也称为染色体计数法)可以使用少到一种bac探针来进行,但是从所述单种探针获得的样本/正常比值的噪音会使得结果不确切。使用靶向受试者染色体的多种探针(其中大多数探针全都表现出相同的获得或丢失)为采用acgh分析的非整倍性检测结果提供了更多的可信度,并且这是cgh分析的常规构建方法。在明确地“称为”或确定为获得或者丢失之前,大部分阵列分析都需要某个区域中的至少两种或三种或更多种bac探针的一致的比值信号。
获得或者丢失区域中额外的个体探针分子会增加获得或者丢失检测的可信度,这在分析信号比有噪音的情况下是有价值的。噪音因为多种原因而存在于分析之中。有时,分析有噪音仅仅是因为探针固定或者样本孵育条件不一致或者是因为标记、杂交、洗涤或干燥中条件并非最佳。在其他情况下,噪音可能是由于使用全基因组扩增(wga)如phi-29或doppcr方法扩增的样本所引起的,其中扩增样本表现出序列特异性扩增偏好。在初始的样本量有限时,例如来自单个细胞或者来自仅几个细胞的dna时,使用wga。这样的扩增样本具有随基因组的不同部分而改变的dna产物产量(扩增偏好),从而将基因组获得-丢失噪音叠加到通常会导致在多种分别排列或分析的bac探针之间形成显著的样本/参照比值反应差异的分析上。补偿这一噪音的标准方法是使用多种探针以跨越目的基因组序列区域,并以某些方式,例如将贯穿基因组区域的比值反应取平均值、利用多种探针的复合结果来作出贯穿该区域的获得-丢失结论。在寡核苷酸cgh阵列中,将多种分别排列或者分析的探针之间的比值取平均值是特别常见的,这通常表现出比bac阵列更大的探针与探针之间的比值差异。
遗憾的是,为了增加可信度而使用针对同一区域的多种分别排列或分析的探针可能受到限制。例如,存在这样的固定探针阵列的布局,其中使用多种分别排列或分析的探针来检测各非整倍性问题重重。一些cgh阵列形式局限于其能容纳的探针数目。所述阵列的一个实例是印在微量板孔底部的阵列,或者印在显微镜用载玻片基底上一个小区域上的阵列,所述基底被构造成可在单个基底上容纳多个独立的样本。这些阵列通常使用9(3×3点阵列矩阵)至100(10×10)种探针。在所述100重的例子中,最多能容纳4种探针来对24条染色体(1-22,x和y)中的每一条进行计数,并且对于染色体计数法而言,阵列的整个容量将被有效地用尽。
除了平面微阵列之外,另一种同时使用多种探针来测量样本中的基因组获得和丢失的方法是使用一系列固定有探针的编码颗粒或微球。使用最为广泛的颗粒平台是luminexxmap系统,所述系统使用荧光颜色编码来区分一百个不同的微球或小球类型。在基因组获得-丢失分析中,该系统可以支持最多达100种不同的探针。同微阵列一样,这一编码微球平台并不支持对由竞争性杂交得到比值的双染双色读数,但是通过对各测试样本和参照样本进行独立的并行分析、进行恰当地标准化并计算比值可以生成相同的比值。
在最大探针数受到局限的这些多重分析形式中,一直都需要对最多24条染色体(1-22,加上x和y)的非整倍性进行可靠地分析、但不为此目的而使用全部或者即使大部分的可能的探针系列。例如,在先天性障碍分析系列中,通常最希望的是不仅要探测非整倍性,还要探测几种微缺失障碍。同样地,使用一种或者两种低分辨力的探针而非更多的常规探针,可以稳定地进行微缺失障碍分析(例如wolf-hirschhorn、willams-beuren、cri-du-chat等),从而在探针受限的平台上分析更多障碍。类似地,在癌症细胞遗传学中,除了所需解析力为整条染色体或者染色体臂丢失的其他区域之外,通常还有几个需要相当高解析力的获得-丢失数据的区域。当仅需要低解析力的基因组区域并不利用系列中相当数量的探针,从而为更多针对需要更高解析力的区域的探针腾出了分析平台容量时,这些系列将更为有用。
因此,需要这样的方法和组合物,其能可靠地分析染色体水平和染色体臂水平的获得和丢失,同时使得可以选择用一种或者多种较高解析力的探针同时地分析其他的基因组区域。
技术实现要素:
本文描述了一种分析dna样本的方法,所述方法包括提供一种与基底相连的复合核酸探针。所述复合核酸探针含有与参照基因组的基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列。所述基因组区域的特征在于具有第一末端和第二末端以及置于所述末端之间的至少400kb的中间区域。所述复合核酸探针包含基本上与含有所述基因组区域第一末端的完整第一基因组座位特异性地杂交的核酸序列,以及基本上与含有所述第二末端的完整第二基因组座位特异性地杂交的核酸序列。所述第一基因组座位和第二基因组座位通常各自含有至少约100kb。任选地,复合探针的核酸序列与所述基因组内的其他座位特异性地杂交。
与基底相连的复合核酸探针在足以实现特异性杂交的严格度下与样本基因组dna杂交。与基底相连的复合核酸探针也与参照基因组dna杂交。
检测指示所述与基底相连的复合核酸探针与所述样本基因组dna特异性杂交的第一信号,同时检测指示所述与基底相连的复合核酸探针与所述参照基因组dna特异性杂交的第二信号。将所述第一信号与所述第二信号进行比较,从而检测所述第一和第二信号之间的差异,所述差异指示所述样本dna和所述参照dna之间的差异。
与参照基因组的基因组区域中的两个或者更多个基因组座位特异性杂交的核酸序列可以来源于两个或者更多个大插入dna载体。例如,所述核酸序列来源于两个或者更多个大插入dna载体,例如细菌人工染色体、酵母人工染色体、人类人工染色体、黏粒、质粒、噬菌粒、噬菌体dna和f黏粒。与参照基因组的基因组区域中的两个或者更多个基因组座位特异性杂交的核酸序列可以来源于分离的染色体和/或分离的染色体片段。
在一个具体的可选方案中,与参照基因组的基因组区域中的两个或者更多个基因组座位特异性杂交的核酸序列为来源于两个或者更多个大插入dna载体、分离的染色体、分离的染色体片段或者这些或者其他核酸来源的组合的扩增子。
在一个具体的可选方案中,基底为多个颗粒如多个编码颗粒。在另一个可选方案中,所述基底为平面基底。
与参照基因组的基因组区域中的两个或者更多个基因组座位特异性杂交的核酸序列各自的长度范围为约20-250,000个核苷酸,包括端值。
本文提供了一种分析样本核酸的方法,所述方法包括提供包含两个或者更多个编码颗粒系列的混合物的多重试剂,所述编码颗粒系列被编码,从而使得每一编码颗粒系列的各个颗粒均可检测地区别于其他每一编码颗粒系列的各个颗粒。所述编码颗粒含有所连接的与参照基因组的至少一个基因组座位特异性杂交的核酸序列,并且至少一个编码颗粒系列含有所连接的复合核酸探针。
所述多重试剂可同时地或者平行地与样本基因组核酸并与参照核酸杂交。
检测指示所述连接的核酸序列与可检测地标记的样本核酸特异性杂交的第一信号以及指示所述连接的核酸序列与可检测地标记的参照核酸特异性杂交的第二信号。然后鉴定所述编码颗粒,从而将颗粒编码情况与所述第一信号或与所述第二信号联系起来。然后比较各个编码颗粒系列的所述第一信号和所述第二信号,并且所述第一和第二信号的差异指示所述样本和所述参照核酸之间的差异。
本文提供了一种用于分析核酸的试剂,所述试剂含有与固体基底相连的第一复合核酸探针。所述第一复合核酸探针包含与参照基因组的基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列。所述基因组区域的特征在于具有第一末端和第二末端以及置于所述第一末端和第二末端之间的至少400kb的中间区域。所述第一复合核酸探针包含基本上与含有所述基因组区域第一末端的完整第一基因组座位特异性杂交的核酸序列,以及基本上与含有所述基因组区域第二末端的完整第二基因组座位特异性杂交的核酸序列。
在一个具体的可选方案中,用于分析核酸的试剂含有至少两种复合探针,并且可以包括更多种复合探针。例如,与固体基底相连的第二复合核酸探针含有与参照基因组的第二基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列。所述第二基因组区域的特征是具有第一末端和第二末端,并且具有置于所述第一末端和第二末端之间的至少400kb的中间区域。所述第二复合核酸探针包含基本上与含有所述第二基因组区域第一末端的完整第一基因组座位、以及基本上与含有所述第二基因组区域第二末端的完整第二基因组座位特异性杂交的核酸序列。
在一个可选方案中,所述固体基底是平面基底。含有一种或者多种复合探针的阵列可以包含在平面基底上。在另一个可选方案中,非复合探针也可以包含在该平面基底上,从而提供一组复合探针和其他探针。
在另一方面,所述固体基底为第一种多个颗粒。第二种复合探针或非复合探针可以与第二种多个颗粒连接。
所述第一种多个颗粒和所述第二种多个颗粒被可区别地编码,用于某些分析类型中。任选地,混合所述第一种多个和第二种多个可区别地编码颗粒,从而提供一种多重分析试剂。连接有额外探针的额外颗粒可以以多重分析形式或者独立分析形式用于本文所描述的核酸分析中。
本文提供了一种制备用于分析dna的与基底相连的复合核酸探针试剂的方法,所述方法包括分离基本上与含有参照基因组的基因组区域的第一末端的完整第一基因组座位特异性杂交的第一核酸序列。所述方法包括分离基本上与含有参照基因组的基因组区域的第二末端的完整第二基因组座位特异性杂交的第二核酸序列。混合所述第一和所述第二核酸序列,从而制备复合探针,并随后将所述复合探针连接到固体基底,从而制备与基底相连的复合核酸探针试剂,用于分析核酸如基因组dna。
在一个具体的可选方案中,所述第一核酸序列分离自第一种大插入载体,所述第二核酸序列分离自第二种大插入载体。例如,所述第一核酸序列分离自第一种bac,所述第二核酸序列分离自第二种bac。
任选地,在混合之前或者之后扩增所述第一和所述第二核酸序列。
具体实施方案提供了利用两种或者多种参照样本的方法。
具体实施方案提供了分析dna样本的方法,所述方法包括:提供与基底相连的核酸探针;使所述与基底相连的核酸探针与获自受试者的样本基因组dna杂交;
使所述与基底相连的核酸探针与第一种参照基因组dna杂交;并使所述与基底相连的核酸探针与第二种参照基因组dna杂交。随后检测指示所述与基底相连的核酸探针与所述样本基因组dna特异性杂交的第一信号、指示所述与基底相连的核酸探针与所述第一种参照基因组dna特异性杂交的第二信号以及指示所述与基底相连的核酸探针与所述第二种参照基因组dna特异性杂交的第三信号。比较所述第一信号和所述第二信号,从而检测所述第一信号和第二信号之间的差异,所述第一和第二信号的差异指示所述样本dna和所述第一种参照dna之间的差异。还比较所述第一信号和所述第三信号,从而检测所述第一和第三信号之间的差异,所述第一和第三信号之间的差异指示所述样本dna和所述第二种参照dna之间的差异。
任选地,根据分析所述dna样本的方法的实施方案,将所述样本dna和所述第一种参照dna之间的差异与所述样本dna和所述第二种参照dna之间的差异进行比较。
在又一个可选方案中,所述第一种参照基因组dna包括男性特异的基因组dna,所述第二种参照基因组dna包括女性特异的基因组dna,从而使得对所述第一和第二信号的差异的比较以及所述第一和第三信号的差异的比较可指示受试者的性别。
在另一个可选方案中,所述第一种参照基因组dna包括第一种病症特异的基因组dna,所述第二种参照基因组dna包括第二种病症特异的基因组dna。因此,对所述第一和第二信号的差异的比较与所述第一和第三信号的差异的比较可指示所述受试者的疾病状态。
一些实施方案提供了这样的方法,其中所述第一种参照基因组dna包括第一种病症特异的基因组dna,所述第二种参照基因组dna包括第二种病症特异的基因组dna,并且对所述第一和第二信号之间的差异的比较以及对所述第一和第三信号之间的差异的比较指示了所述受试者的新陈代谢年龄。
所述与基底相连的核酸探针可以包含多个编码颗粒和/或一个平面基底。任选地,所述与基底相连的核酸探针包括多种寡核苷酸和/或多种扩增子。所述与基底相连的核酸探针可以包括分离自大插入dna载体和/或分离的染色体dna的插入dna。
本发明的实施方案提供了分析dna样本的方法,所述方法包括:提供含有连接有扩增子的编码颗粒的第一编码颗粒系列。所述扩增子包含合在一起基本上代表完整第一模板dna序列的随机核酸序列。使所述第一编码颗粒系列的扩增子与可检测地标记的样本dna、可检测地标记的第一种参照dna以及可检测地标记的第二种参照dna杂交。检测指示所述第一编码颗粒系列的扩增子与可检测地标记的样本dna特异性杂交的第一信号。检测指示所述第一编码颗粒系列的扩增子与可检测地标记的第一种参照dna特异性杂交的第二信号。检测指示所述第一编码颗粒系列的扩增子与可检测地标记的第二种参照dna特异性杂交的第三信号。比较所述第一信号和所述第二信号,以检测所述第一和第二信号之间的差异,所述第一和第二信号的差异指示所述样本dna和所述参照dna之间的差异。比较所述第一信号和所述第三信号,以检测所述第一和第三信号之间的差异,所述第一和第三信号的差异指示所述样本dna和所述第二种参照dna之间的差异。
在一个任选方案中,所述扩增子的长度范围为大约500-1200个核苷酸,包括端值。
具体实施方案的方法还包括提供含有连接有扩增子的编码颗粒的第二编码颗粒系列,其中所述扩增子包含合在一起基本上代表完整第二模板dna序列的随机核酸序列。使所述第二编码颗粒系列的扩增子与可检测地标记的样本dna、可检测地标记的第一种参照dna和可检测地标记的第二种参照dna杂交。检测指示所述第二编码颗粒系列的扩增子与可检测地标记的样本dna特异性杂交的第一信号。检测指示所述第二编码颗粒系列的扩增子与可检测地标记的第一种参照dna特异性杂交的第二信号。检测指示所述第二编码颗粒系列的扩增子与可检测地标记的第二种参照dna特异性杂交的第三信号。比较所述指示所述第二编码颗粒系列的扩增子的特异性杂交的第一信号和所述指示所述第二编码颗粒系列的扩增子的特异性杂交的第二信号,以检测所述第一信号和第二信号之间的差异,所述第一和第二信号的差异指示所述样本dna和所述第一种参照dna之间的差异。比较所述指示所述第二编码颗粒系列的扩增子的特异性杂交的第一信号和所述指示所述第二编码颗粒系列的扩增子的特异性杂交的第三信号,以检测所述第一和第三信号之间的差异,所述第一和第三信号的差异指示所述样本dna和所述第二种参照dna之间的差异。
任选地,所述第一和第二编码颗粒系列以混合物的形式提供。检测颗粒编码情况,从而将各颗粒系列的身份与通过样本和参照dna杂交获得的具体信号联系起来。
附图说明
图1是示出一个实施方案的流程图,该实施方案包括使用两个扩增反应由模板dna制备扩增子,并将扩增子作为探针固定于一系列编码小球上,其中该系列中的小球均具有相同的id代码;
图1a是示出一个实施方案的流程图,该实施方案包括由单个bac克隆制备bac扩增子,并将该扩增子作为探针固定到一系列编码小球上,从而形成一个小球系列,其中该系列中的小球全都具有相同的id代码;
图2是示出一个实施方案的流程图,所述实施方案包括混合m个不同的编码小球系列,各系列均具有其各自被固定的bac-扩增子探针dna,它们合在一起形成多重编码小球系列;
图3是示出一个实施方案的流程图,所述实施方案包括使用多重编码小球系列在n个样本上进行多重基因组获得和丢失分析;
图3a为示出一个实施方案的流程图,所述实施方案包括使用多重编码小球系列在n个样本上进行多重基因组获得和丢失分析;
图4为sbs标准的96孔微孔板的模式图,其示出了平行地对46份样本运行分析的两份对照和两份样本的示例性位置;
图5为使用男性的13号染色体上具有三体性的corielldna样本所形成的数据的一个实例;
图6为使用男性的18号染色体上具有三体性的corielldna样本所形成的数据的一个实例;
图7为使用女性的21号染色体上具有三体性的corielldna样本所形成的数据的一个实例;
图8为使用具有x染色体5拷贝扩增的corielldna样本所形成的数据的一个实例;
图9为一张图表,其示出了用于在示例性分析中形成固定到编码小球上的扩增子的bac克隆、其染色体和cytoband位置、阴性对照寡核苷酸的序列和固定有各扩增子探针的小球系列的小球id(luminex小球区);
图10a是示出根据本文所述方法的一个方面的一种制备复合探针的方法的简化流程图;
图10b是示出根据本文所述方法的一个方面的一种制备复合探针的方法的简化流程图;
图11是示出根据本文所述方法的一个方面的一种制备复合探针的方法的简化流程图;
图12是由测试分析获得的数据绘制的图,示出了复合探针与digeorge综合征参照dna样本的使用;
图13是一幅示出由使用两种参照基因组dna样本的luminex小球阵列获得-丢失分析得到的比值数据的图;和
图14是一幅示出由使用两种参照基因组dna样本的luminex小球阵列获得-丢失分析得到的比值数据的图。
具体实施方式
本文提供了与染色体获得和丢失分析相关的方法和组合物。泛泛的说,本文描述了与使用与基底相连的核酸探针的基因组dna获得和丢失分析相关的方法和组合物。
本文使用的科技术语旨在具有本领域普通技术人员通常所理解的含义。这些术语在多部权威的参考书中被定义和使用,所述参考书例如包括j.sambrookandd.w.russell,molecularcloning:alaboratorymanual,coldspringharborlaboratorypress;3rded.,2001;f.m.ausubel,ed.,shortprotocolsinmolecularbiology,currentprotocols;5thed.,2002;b.albertsetal.,molecularbiologyofthecell,4thed.,garland,2002;d.l.nelsonandm.m.cox,lehningerprinciplesofbiochemistry,4thed.,w.h.freeman&company,2004;和herdewijn,p.(ed.),oligonulceotidesynthesis:methodsandapplications,methodsinmolecularbiology,humanapress,2004。
本文所使用的术语“核酸”是指任何形式(包括单链、双链的寡核苷酸或多核苷酸)的具有多于1个核苷酸的rna或dna分子。
探针
本文所使用的术语“探针”是指用于识别靶核酸的核酸,所述靶核酸与所述探针特异性结合。
用于本文所述分析中的核酸探针可以包括细胞或者生物体的基因组的全部的或者部分。所述核酸探针可以包括表现为一条或者多条染色体、染色体的一部分、基因座、基因或基因的一部分的dna。所述核酸探针可以为任何形式如载体中的插入物,所述载体例如包括细菌人工染色体、酵母人工染色体、人类人工染色体、黏粒、质粒、噬菌粒、噬菌体dna或f黏粒。所述核酸探针可以是显微切割的染色体dna形式。因此,尽管本文描述的具体实例是作为核酸探针dna来源的bac,但是也可以使用其他类型的克隆,如pac、yac、黏粒、f黏粒、cdna等。
在具体应用中,核酸被用作用于扩增的模板材料,所得到的扩增子或其部分被用作探针。
用于形成核酸探针、样本或者参照的核酸均通过本领域已知方法获得,例如如j.sambrookandd.w.russell,molecularcloning:alaboratorymanual,coldspringharborlaboratorypress;3rded.,2001或f.m.ausubel,ed.,shortprotocolsinmolecularbiology,currentprotocols;5thed.,2002所述。核酸还可以购买获得和/或使用市售试剂盒获得。
复合探针
具体实施方案提供了一种复合核酸探针,所述复合核酸探针含有与参照基因组中目标基因组区域的两个或者更多个基因组座位特异性杂交的核酸序列。所述基因组区域的特征是具有第一末端和第二末端以及置于所述末端之间的至少400kb的中间区域。复合核酸探针包含基本上与含有基因组区域第一末端的完整第一基因组座位特异性杂交的核酸序列,以及基本上与含有第二末端的完整第二基因组座位特异性地杂交的核酸序列。第一基因组座位和第二基因组座位通常分别都含有至少大约100kb,并且可以更大。
与参照基因组的基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列可以来源于两个或者更多个大插入dna载体。例如,所述核酸序列来源于两个或者更多个大插入dna载体,如细菌人工染色体、酵母人工染色体、人类人工染色体、pl来源的人工染色体(pac)、黏粒、质粒、噬菌粒、噬菌体dna和f黏粒。与参照基因组的基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列也可以来源于分离的染色体和/或分离的染色体片段。
在一个具体的可选方案中,与参照基因组的基因组座位中两个或者更多个基因组座位特异性杂交的核酸序列为由来源于两个或者更多个大插入dna载体、分离的染色体、分离的染色体片段或者这些或其他核酸来源的组合的模板扩增得到的扩增子。
在具体实施方案中,与参照基因组的基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列以寡核苷酸和/或多核苷酸的形式提供,其各自的长度范围为大约20-250,000个核苷酸,包括端值,并且合在一起基本上可与含有所述基因组区域第一末端的完整第一基因组座位特异性杂交,并基本上与含有所述基因组区域第二末端的完整第二基因组座位特异性杂交。
因此,复合探针可以含有2种或更多种bac的汇集物(pool),或者来源于2种或更多种bac的插入dna。任选地,所述汇集物可以含有来源于4种至约100种bac(包括端值)的插入dna。所述探针dna可以提取自经培养的bac,或者在一个具体的实施方案中,可以为利用例如简并寡核苷酸引物(dop)pcr或连接反应介导的pcr由bacdna获得的扩增子。可以使用其他大插入物克隆如pac、yac、黏粒、f黏粒等来替代bac。也可以使用大量寡核苷酸的汇集物。
在一个具体的方面,固定的复合探针可以是来源于经分选的染色体的dna。可以使用分选细胞的流式细胞仪(也称为荧光激活的细胞分选仪;facs)来形成经分选染色体的汇集物,其中所述分选以对基因组的at富集区和gc富集区特异的染料之间的信号比为基础。由所述经分选染色体提取的dna可以通过wga扩增,并用荧光染料标记,以在中期染色体fish分析中用作染色体涂染探针。类似地,染色体臂涂染探针也可以通过以下方式制备:对利用激光捕获显微切割从中期染色体展片(metaphasespread)分离的经分选染色体臂的dna进行扩增。以微点阵形式固定或者固定在编码颗粒上的固定全染色体或染色体臂探针产生分别代表跨越所述全染色体或臂的平均获得或者丢失情况的杂交比信号,其方式与跨越整个染色体或臂的颇为大量的bac汇集物相似。
复合探针在多重基因组获得-丢失分析(例如本文所述的多重基因组获得-丢失分析)中起到多种作用。它们包括染色体计数(非整倍性检测)、微缺失或其他综合征的检测或者作为对照。将复合探针用作对照来确定正常的常染色体应答(当前样本/参照比值的水平=1.0)可以对较大跨度的基因组上的应答取平均,从而可以降低个体之间的正常拷贝数差异所引起的比值噪音。
制备复合探针的方法
本文提供了一种制备与基底相连的用于分析dna的复合核酸探针试剂的方法。分离基本上与含有参照基因组的基因组区域第一末端的完整第一基因组座位特异性杂交的第一核酸序列。另外,分离至少一种基本上与含有所述参照基因组dna的所述基因组区域第二末端的完整第二基因组座位特异性杂交的第二核酸序列。术语“分离”在用于核酸时是指核酸与同其天然一起存在的其他物质(如细胞、蛋白和其他核酸)基本分离。混合所述第一和所述第二核酸序列,从而制备复合探针,并在随后将所述复合探针连接到固体基底,从而制备与基底相连的复合核酸探针试剂,用于分析核酸如基因组dna。
在一个具体的可选方案中,所述第一核酸序列分离自第一种大插入载体,所述第二核酸序列分离自第二种大插入载体。例如,所述第一核酸序列为分离自第一种bac的dna插入物,所述第二核酸序列为分离自第二种bac的dna插入物。
在又一个可选方案中,所述第一核酸序列为分离自第一种bac的人类基因组dna插入物,所述第二核酸序列为分离自第二种bac的人类基因组dna插入物。
任选地,在混合之前或者之后扩增所述核酸序列。例如,在混合之前或者之后扩增所述第一和所述第二或者更多的核酸序列。
由复合探针中核酸序列靶向的基因组座位的数目并不局限于两种,可以为三种、四种或更多种,如大约4-100种或更多种。因此,同样地,与基因组座位特异性杂交的具体核酸序列的数目也不局限于两种,可以为三种、四种或更多种,如大约4-100种或更多种。
复合探针的一个实例是使用来自五种bac的插入dna制备的复合探针,所述插入dna被定位到与digeorge微缺失综合征相对应的cytoband22p11.2。所用五种bac的中心座位跨越大约0.45mb(445kb),对于175kb的典型bac长度,所述总跨度略高于600kb。对从所述五种bac中每一种分离的插入dna进行扩增,并将所得到的扩增子汇集起来制备复合探针。随后将所述复合探针连接到一个系列的luminex编码多重微球,所述微球全都具有相同的小球编码身份,用于在多重基因组获得-丢失分析中用作22p11.2cytoband探针。
因此,在一个具体的实例中,2种、3种、4种、5种或更多种,(如6-100种或更多种)来自bac或其他大插入载体的插入物,可以用作与确定基因组区域中具体基因组座位特异性杂交的核酸序列。
基底
用于连接探针(包括复合探针)的固体基底(包括半固体基底),可以为多种材料中的任何一种,如玻璃;塑料,如聚丙烯、聚苯乙烯、尼龙;纸;硅;硝酸纤维素;或核酸可以与之连接以用于进行分析的任何其他材料。所述基底可以为多种形式或者形状中的任何一种,包括平面的(如硅片和玻璃板)和三维的(如颗粒、微量滴定板、微量滴定孔、针、纤维等)。
在具体的方面,与探针相连的固体基底为颗粒。
与探针相连的颗粒可以为任何探针可与之相连的固体或半固体颗粒,该颗粒适合核酸杂交分析,并且在杂交和检测条件下是稳定的且不溶的。所述颗粒可以为任何形状(如圆柱形、球形等)、尺寸、组成或具有任何理化特征。可以对粒度或组成进行选择,从而使得颗粒可以在例如具有特定孔径的过滤器上或者通过一些其他的物理性质(如磁性)与流体相分离。
所使用的微粒如微球的直径可以小于一毫米,例如,直径的尺寸范围为约0.1至约1,000微米(包括端值),如直径为约3-25微米(包括端值),或者直径为约5-10微米(包括端值)。所使用的纳米颗粒如纳米小球的直径可为约1纳米(nm)至约100,000nm(包括端值),例如,尺寸范围为约10-1,000nm(包括端值),或者例如,尺寸范围为200-500nm(包括端值)。在某些实施方案中,所使用的颗粒为小球,特别是微球和纳米小球。
举例来说,颗粒为有机或无机颗粒,如玻璃或金属颗粒,并且可以为合成的或者天然的聚合物的颗粒,所述聚合物如聚苯乙烯、聚碳酸酯、聚硅酮(silicon)、尼龙、纤维素、琼脂糖、葡聚糖和聚丙烯酰胺。在具体的实施方案中,颗粒为乳胶小球。
在具体的实施方案中,所使用的颗粒含有用于连接核酸的官能团。例如,颗粒可以含有羰基、胺、氨基、羧酸基团、卤素、酯、醇、脲、醛、氯甲基、氧化硫、一氧化氮、环氧基和/或甲苯磺酰基官能团。颗粒的官能团、对其的修饰及其与化学部分(如核酸)的连接为本领域所熟知,例如如fitch,r.m.,polymercolloids:acomprehensiveintroduction,academicpress,1997中所述。美国专利no.6,048,695描述了一种用于将核酸探针如扩增子连接到基底如颗粒上的示例性方法。在另一个具体的实例中,1-乙基-3-[3-二甲基氨丙基]碳化二亚胺盐酸盐(edc或edac化学品)可以用来将核酸连接到编码颗粒。
编码颗粒为基于某个特征可以与其他颗粒区分开的颗粒,所述特征例如包括光学性质如颜色、反射指数和/或压印图案或光学可检测的图案。例如,可以使用光学标签、化学标签、物理标签或者电学标签来编码颗粒。编码颗粒可以含有一个或者多个荧光团或与之相连,所述荧光团可以通过例如激发和/或发射波长、发射强度、激发态寿命或这些的组合、或者其他光学特征进行区分。可以使用光学条形码来对颗粒进行编码。
在具体的实施方案中,一个颗粒系列的各颗粒均用相同的代码进行编码,从而使得一个颗粒系列的各颗粒都可以与另一个颗粒系列的各颗粒区分开。在其他实施方案中,可以对单个颗粒系列使用两种或者多种代码。例如,各颗粒都可以含有独特的代码。在某些实施方案中,颗粒编码包含一个代码,但不包含颗粒与对基因组dna特异的核酸探针之间的联系,或者兼而有之。
在具体的实施方案中,代码镶嵌在例如颗粒内部,或者以在杂交和分析期间稳定存在的方式与颗粒相连。可通过任何可检测的方式提供代码,如通过全息编码,通过荧光性质、颜色、形状、尺寸、光发射、量子点发射等,以鉴定颗粒,并由此鉴定固定于其上的捕获探针。在一些实施方案中,代码不是由核酸提供。
一个示例性平台利用浸渍于聚合物颗粒内的荧光染料的混合物,作为鉴定固定有特定俘获探针的颗粒系列的每个成员的工具。另一个示例性平台使用全息条码来鉴定圆柱形玻璃颗粒。例如,chandler等人(美国专利no.5,981,180)描述了一种基于颗粒的系统,其中不同的颗粒类型由不同比例的两种或更多种浸渍于聚合物颗粒内的荧光染料的混合物编码。soini(美国专利no.5,028,545)描述了一种基于颗粒的多重分析系统,该系统采用时间分辨荧光进行颗粒鉴定。fulwyler(美国专利no.4,499,052)描述了一种使用通过颜色和/或尺寸区分的颗粒的示例性方法。美国专利申请公开文本20040179267、20040132205、20040130786、20040130761、20040126875、20040125424和20040075907描述了由全息条码编码的示例性颗粒。美国专利no.6,916,661描述了与纳米颗粒有关的聚合物微粒,所述纳米颗粒具有为颗粒提供代码的染料。
尽管本文详细描述的一个实施方案利用的是luminex编码小球平台,但是也可以使用其他类型的编码颗粒分析平台,如veracode小球和beadxpress系统(illuminainc.,sandiegoca)、xmap3d(luminex)等。可以使用磁性luminex小球,该小球使得可以使用板状磁铁和移液管而不使用过滤板和多头抽真空装置进行洗涤步骤。这其中的每一个平台通常都以羰基小球的形式提供,但是也可以对其进行构建,从而使其含有不同的偶联化学组成,如氨基-硅烷。
与基底的连接
核酸探针与基底的连接可以通过将核酸有效地键合到固体或半固体基底的多种方法中的任何一种实现,举例说来,所述方法包括吸附和化学键合。核酸可以直接地键合至所述编码颗粒的材料,或者间接地(例如通过键合至分布于颗粒之上的包衣或接头)键合至所述编码颗粒。可以合成核酸,并且/或者在合成后对其进行修饰,从而使其含有用于将所述核酸键合至颗粒的官能团。例如,用作探针的核酸序列可以含有羰基、胺、氨基、羧酸基团、卤素、酯、醇、脲、醛、氯甲基、氧化硫、一氧化氮、环氧基和/或甲苯磺酰基官能团。
与基底相连的探针(包括复合探针)可以为单链的和/或双链的核酸。在具体实施方案中,在双链核酸被连接时,在将其固定于基底之后使其变性成为单链以进行制备,从而用于分析方法的某些实施方案中。任选地,在固定之前使双链核酸探针变性,然后将单链的核酸连接至基底。
扩增子试剂组合物
在具体实施方案中,提供了一种用于分析核酸的试剂,所述试剂包含连接有作为探针的扩增子的多个编码颗粒。
在其他具体实施方案中,所连接的扩增子由多于一种模板扩增得到,从而形成了复合探针。
在某些实施方案中,与多个编码颗粒连接的扩增子每一个都含有与模板基因组核酸的一部分相同或者完全互补的核酸序列,并且所述扩增子合在一起基本上代表完整的模板基因组核酸。
图1示出一种制备用于分析基因组dna的试剂的方法的一个实施方案。如图1所示,提供了一个模板核酸(1)。使用简并寡核苷酸引物(dop)在第一扩增反应中扩增所述模板(2),从而产生第一扩增产物。
所述模板核酸可以是能够使用核酸扩增方法被复制的任何核酸。
用于所述第一扩增反应的模板dna任选地为基因组dna,其大小范围通常为约20-300kb,但是该模板也可以更小或者更大。术语“基因组的”指细胞或者生物体的基因组的dna,其不仅包括由细胞或者生物体的基因组的dna复制得到的dna(如克隆得到的dna),还包括从细胞或生物体直接分离得到的dna(如显微切割的染色体dna)。模板dna可以包括细胞或者生物体的全部的或者部分的基因组。模板dna可以包括表现为一种或者多种染色体、染色体的一部分、基因座、基因或基因的一部分的dna。模板dna可以为任何形式,如载体中的插入物,所述载体例如包括细菌人工染色体、酵母人工染色体、人类人工染色体、黏粒、质粒、噬菌粒、噬菌体dna或f黏粒。模板dna可以是显微切割的染色体dna形式。因此,尽管本文描述的具体实例是以bac作为模板dna来源,但是也可以使用其他类型的克隆,如pac、yac、黏粒、f黏粒、cdna等。
根据具体的实施方案,可以一起或者分别地扩增来自于这些或者其他来源中任何一种的多个模板,合并得到的来自所述多个模板的扩增子为复合探针。
模板基因组dna通过本领域已知的方法获得,例如描述于以下文献中的方法:j.sambrookandd.w.russell,molecularcloning:alaboratorymanual,coldspringharborlaboratorypress;3rded.,2001,或者f.m.ausubel,ed.,shortprotocolsinmolecularbiology,currentprotocols;5thed.,2002。模板dna还可以购买获得和/或使用分离基因组dna的商用试剂盒获得。
使用体外扩增方法可以实现对模板dna的扩增。术语“扩增方法”指这样一种方法或技术,即所述方法或技术用于复制模板核酸,由此产生包括所述模板核酸的全部或者部分的多个拷贝的核酸,所产生的核酸也称为扩增子。
扩增子任选地含有存在于引物中且不存在于原始dna模板中的核酸序列。所述引物来源的核酸增加了功能性,例如用于其他扩增反应的引物结合位点,和/或与基底进行化学键合的官能团。
举例而言,扩增方法包括pcr、连接反应介导的pcr(lm-pcr)、phi-29pcr和其他核酸扩增方法,例如如c.w.dieffenbachetal.,pcrprimer:alaboratorymanual,coldspringharborlaboratorypress,2003;和v.demidovetal.,dnaamplification:currenttechnologiesandapplications,taylor&francis,2004中所述。
可以使用具体的dna模板来源和核酸扩增方法的多种结合。
术语“寡核苷酸引物”指能够在合适的反应条件下担当引物延伸产物的合成起始位点的核酸。寡核苷酸引物的长度通常为约10–30个连续的核苷酸。寡核苷酸引物与模板核酸的一个区域完全互补或者基本互补,因而在杂交条件下,所述寡核苷酸引物与模板核酸的所述互补区域退火。引物延伸产物合成的合适反应条件包括存在合适的反应组分(包括但不限于聚合酶和核苷三磷酸)。对于适合用于扩增反应中的寡核苷酸引物的设计为本领域所熟知,例如如a.yuryevetal.,pcrprimerdesign,humanapress,2007中所述。
术语“简并寡核苷酸引物”指含有具有随机或者半随机核苷酸序列的核酸的引物。对于适合用于具体核酸扩增反应的简并寡核苷酸引物的设计为本领域所熟知,例如如a.yuryevetal.,pcrprimerdesign,humanapress,2007中所述。可以使用具有大约5-8个核苷酸的随机或半随机核苷酸序列。在其他实施方案中,随机或半随机核苷酸六聚体包含在用于第一扩增反应的简并寡核苷酸引物中。
在具体的实施方案中所使用的简并寡核苷酸引物每一个均包含一个5'恒定dna区段、一个中间随机dna区段和一个3'锚定区段,例如如fiegleretal.,geneschromosomescancer,36(4):361-74,2003;和telenius,etal.,genomics13:718-25,1992中所述。5'恒定dna区段任选地具有在所有dop中均相同的核苷酸序列。3'锚定区段任选地具有经测定在模板核酸中具有所需出现频率的核苷酸序列。对具体的核酸序列出现频率的分析为本领域所熟知,例如如milosavljic,a.andjurka,j.,1993,comput.applic.biosci.,9:407-411;pesole,g.etal.,1992,nucleicacids,res.,20:2871-2875;和hutchinson,g.b.,1996,comput.appl.biosci.,12:391-398中所述。
在具体的实施方案中,dop含有约17-25个连续的核苷酸,其中约7-12个连续的核苷酸包含于5'恒定dna区段之中,约5-8个连续的核苷酸包含于随机dna区段之中,约5-8个连续的核苷酸包含于3'锚定区段之中。
第一扩增反应产生含有多个扩增子的第一反应产物。第一反应产物中的各扩增子均含有与dna模板的一个随机部分相同或者完全互补的一段dna序列以及与第一种反应引物的5'恒定dna序列相同的一段dna序列。
本文中使用的术语“互补的”是指核苷酸之间的沃森-克里克碱基配对,具体地指核苷酸通过氢键彼此键合,胸腺嘧啶或尿嘧啶残基通过两个氢键与腺嘌呤残基相连,而胞嘧啶和鸟嘌呤残基通过三个氢键相连。通常,核酸所包含的核苷酸序列被描述为与指定的第二核苷酸序列具有某一“百分比的互补度”。例如,一段核苷酸序列可以与指定的第二核苷酸序列具有80%、90%或100%的互补度,这表明一段10个核苷酸的序列中有8个、9个或者10个核苷酸与指定的第二核苷酸序列互补。例如,核苷酸序列3'-tcga-5'与核苷酸序列5'-agct-3'100%互补。另外,核苷酸序列3'-tcga-与核苷酸序列5'-ttagctgg-3'的一个区域100%互补或完全互补。
参照图1,使用第一反应产物扩增子作为模板dna进行第二扩增反应(3)。第二扩增反应(3)包括“通用”寡核苷酸引物,之所以这样称谓是因为该通用引物与在第一扩增反应中使用的dop的5'恒定dna区段相同或者完全互补。通用寡核苷酸引物包含在第一扩增反应中使用的dop的5'恒定dna区段,所述5'恒定dna区段位于该通用引物的3'末端。通用寡核苷酸引物任选地在其5’末端含有其他连续的核苷酸。
在一个具体的可选方案中,在通用寡核苷酸引物的5’端含有一个官能团,用于将由第二扩增反应产生的扩增子连接至编码固体或半固体基底如编码颗粒。例如,在通用寡核苷酸引物的5’端含有一个胺基基。在另一个可选方案中,可以对由第二扩增反应产生的扩增子进行修饰,从而使其含有用于键合至固体或者半固体基底的官能团。修饰核酸从而使其含有能够键合至固体或者半固体基底的官能团为本领域所熟知。
在一个具体的实施方案中,与颗粒相连的每一个单独的扩增子均含有与模板dna序列的一个随机部分相同的一个dna区段。每一个单独的扩增子均还含有一个恒定dna区段,所述恒定dna区段同与模板dna序列的一个随机部分相同的dna区段毗邻。扩增子的恒定dna区段任选地含有一个末端官能团,用于将扩增子连接到编码颗粒。在一个具体的实施方案中,所述扩增子的恒定dna区段含有一个5'末端胺基团,用于将扩增子连接到编码颗粒。
如图1所示,将第二反应产物的扩增子固定于第一种多个编码颗粒上(4)。第二扩增反应的扩增子与所述编码颗粒的连接可通过多种可有效地键合核酸与固体或者半固定基底的方法之一实现,所述方法例如包括吸附和化学键合。扩增子可以与编码颗粒的材料直接键合,或者与编码颗粒间接键合,例如,通过键合至分布于颗粒上的包衣或接头。可以合成扩增子,并且/或者在合成后对其进行修饰,从而使其含有一个用于将扩增子键合到颗粒上的官能团。例如,扩增子可以含有羰基、胺、氨基、羧酸基团、卤素、酯、醇、脲、醛、氯甲基、氧化硫、一氧化氮、环氧基和/或甲苯磺酰基官能团。
通常,作为第二扩增反应的产物的扩增子是双链的,并且该双链的扩增子与颗粒相连。因此,所述双链扩增子的两条链都呈现于每一个颗粒之上。在将扩增子固定于颗粒之后使其变性成为单链,以进行制备,从而用于分析方法的具体实施方案中。任选地,在固定之前使双链扩增子变性,然后将单链的扩增子与颗粒相连。
如所述,第一和第二扩增反应两者中的每一个扩增子均含有与模板dna序列的一个随机部分相同的一段核酸序列,从而使由第一扩增反应产生的扩增子合在一起基本上代表完整的模板dna序列,由第二扩增反应产生的扩增子合在一起基本上代表完整的模板dna序列。
用于分析基因组dna的第一颗粒系列和第一试剂为连接有扩增子的编码颗粒,所述扩增子是第二扩增反应的产物,其合在一起基本上代表在第一扩增反应中用作模板的完整基因组dna序列。
在具体的实施方案中,与颗粒相连的每一个单独的扩增子的长度范围为约500–1200个核苷酸(包括端值)。因此,通过所连接的由所述模板扩增得到的相对较小的扩增子,相对较大的模板核酸基本完整地呈现于一个系列的编码颗粒之上。
如上文所述,各颗粒系列均含有连接有扩增子的编码颗粒,所述扩增子是第二扩增反应的产物,其合在一起基本上代表在第一扩增反应中用作模板的完整基因组dna序列。含有扩增子的颗粒数目取决于诸多因素如模板尺寸、扩增子尺寸以及可用于将扩增子连接到颗粒之上的结合位点的数目,所述含有扩增子的颗粒数目足以使得合在一起基本上代表在第一扩增反应中用作模板的完整基因组dna序列。通常,足以使得合在一起基本上代表在第一扩增反应中用作模板的完整基因组dna序列的颗粒数目的范围为约1-10,000(包括端值)。
使用第二种基因组dna模板进行扩增并将作为第二扩增反应的产物的扩增子(如上所述)连接至第二种多个编码颗粒,形成了另外的颗粒系列。所述第二种多个编码颗粒可检测地不同于所述第一种多个编码颗粒,从而形成用于分析基因组dna的第二编码颗粒系列和第二试剂。
类似地,将第三种或后续的基因组dna模板用于形成扩增反应的反应产物,并将该反应产物连接至第三种或后续的多个编码颗粒。所述第三种或后续的多个编码颗粒中的每一种均可检测地不同于其他的每一种多个编码颗粒,从而获得用于分析基因组dna的第三或后续的编码颗粒系列和第三或后续的试剂。
多重试剂
某些实施方案提供了一种用于分析基因组dna的多重试剂,所述多重试剂包含两个或更多个颗粒系列的混合物。在具体的实施方案中,每一编码颗粒系列的各个编码颗粒均可检测地区别于其他每一编码颗粒系列的各个编码颗粒。
在具体的实施方案中,至少一个颗粒系列包含复合探针。任选地,多于一个颗粒系列包含复合探针。
在具体的实施方案中,每一编码颗粒系列均连接有扩增子,所述扩增子为如本文所述的第二扩增反应的产物,并且合在一起基本上代表在第一扩增反应中用作模板的完整基因组dna序列,其中不同的基因组模板由与其他各编码颗粒系列相连的扩增子所代表。
一个具体实施方案的一种多重试剂包括连接有扩增子的第一编码颗粒系列和连接有扩增子的第二编码颗粒系列,所述与第一编码颗粒系列相连的扩增子合在一起基本上代表插入第一细菌人工染色体中的完整模板dna序列,而所述与第二编码颗粒系列相连的扩增子合在一起基本上代表插入第二细菌人工染色体中的完整模板dna序列。
例如,与第一编码颗粒系列相连的扩增子含有与人13号染色体dna的一部分相同的核酸序列,而与第二编码颗粒系列相连的扩增子含有与人18号染色体dna的一部分相同的核酸序列。与第三或后续的编码颗粒系列相连的扩增子含有与人的另一染色体或染色体的另一非重叠区域的dna相同的核酸序列。
本文所描述的多重试剂可以在单个分析中同时分析多个靶标,如多个基因组座位(genomicloci)。
通过至少混合第一编码颗粒系列和第二编码颗粒系列形成了用于分析基因组dna的多重试剂。
图2示出了一种形成多重试剂的方法的一个实施方案。如图所示,任何数目“m”的编码颗粒系列均可以包含在所述多重试剂中。因此,例如,“m”可以为至少2、3、4、5、10、15、20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100或200个不同的编码颗粒系列。一个系列的连接有扩增子的编码颗粒与一个或者多个其他系列的连接有扩增子的编码颗粒联合形成用于分析样本中基因组获得和丢失的多重试剂。
在一个具体的实施方案中,包含结合的复合探针的一个编码颗粒系列与具有结合的复合探针或非复合探针的一个或多个其他编码颗粒系列相联合,形成用于分析样本中基因组获得和丢失的多重试剂。
分析方法
具体实施方案提供了一种分析dna样本的方法,所述方法包括提供一种与基底相连的复合核酸探针。所述复合核酸探针含有与参照基因组的基因组区域中两个或者更多个基因组座位特异性杂交的核酸序列。所述基因组区域的特征在于具有第一末端和第二末端以及介于所述末端之间的至少400kb的中间区域。所述复合核酸探针包含基本上与含有所述基因组区域第一末端的完整第一基因组座位特异性杂交的核酸序列,以及基本上与含有第二末端的完整第二基因组座位特异性杂交的核酸序列。所述第一基因组座位和第二基因组座位各自通常含有至少大约100kb。任选地,复合探针的核酸序列与所述基因组区域中的其他座位特异性杂交。在又一个可选方案中,参照基因组为一个或者多个人类基因组。
与所述基底相连的复合核酸探针在足以实现特异性杂交的严格度下与样本基因组dna杂交。所述与基底相连的复合核酸探针还在相同或者相似的条件下与参照基因组dna杂交,从而可以进行比较。
检测指示所述与基底相连的复合核酸探针与所述样本基因组dna特异性杂交的第一信号,以及指示所述与基底相连的复合核酸探针与所述参照基因组dna特异性杂交的第二信号。比较所述第一信号和第二信号,从而检测所述第一和第二信号之间的差异,该差异指示所述样本dna和所述参照dna之间的差异。
分析样本核酸的方法的具体实施方案包括提供含有两个或者更多个编码颗粒系列的混合物的多重试剂,所述编码颗粒系列被编码,从而使每一编码颗粒系列的各颗粒可检测地区别于其他每一编码颗粒系列的各颗粒。所述编码颗粒包含所连接的与参照基因组的至少一个基因组座位特异性杂交的核酸序列,并且至少一个编码颗粒系列含有连接的复合核酸探针。
所述多重试剂同时地或者平行地与样本基因组核酸和参照核酸杂交。
检测指示所连接的核酸序列与可检测地标记的样本核酸特异性杂交的第一信号,并检测指示所连接的核酸序列与可检测地标记的参照核酸特异性杂交的第二信号。然后鉴定所述编码颗粒,从而将颗粒编码情况与所述第一信号或者与所述第二信号联系起来。随后比较各个编码颗粒系列的第一信号和第二信号,所述第一和第二信号的差异指示所述样本和参照核酸之间的差异。
在具体的实施方案中,分析基因组dna的方法包括提供连接有扩增子的编码颗粒,所述扩增子合在一起基本上代表完整的模板基因组核酸。在具体的实施方案中,提供了连接有扩增子的编码颗粒,所述扩增子合在一起基本上代表不止一个拷贝的完整的模板基因组核酸。
在具体的实施方案中,准备用于分析基因组获得和/或丢失的基因组dna样本用可检测标记进行标记。参照dna也用可检测标记进行标记,用于与样本dna进行比较。样本和参照dna可以用相同的或者不同的可检测标记进行标记,这取决于所使用的分析安排。例如,在具体的实施方案中,用不同的可检测标记所标记的样本和参照dna可以在同一容器中一起使用,用于同与编码颗粒相连的扩增子杂交。在其他实施方案中,用相同的可检测标记所标记的样本和参照dna可以在分开的容器中使用,用于同与颗粒相连的扩增子杂交。
术语“可检测标记”指可提供可检测信号并且可以与核酸连接的任何原子或部分(moiety)。所述可检测标记的实例包括荧光部分、化学发光部分、生物发光部分、配体、磁性颗粒、酶、酶底物、放射性同位素和生色团。
多种标记样本和参照核酸如dna的方法中的任何一种方法均可以用于本分析中,如核酸的缺口平移或化学标记。例如,通过使用如下的核苷酸进行聚合反应可以引入可检测标记,所述核苷酸包括至少一些经修饰的核苷酸,如经修饰后含有生物素、地高辛、荧光素或花菁的核苷酸。在一些实施方案中,通过随机引物法和聚合反应引入可检测标记。其他实例包括缺口平移(rocheappliedscience,indianapolisind.;invitrogen,carlsbadcalif.)和化学标记(kreatechuls,amsterdamnl)。对核酸的可检测标记为本领域所熟知,并且可以使用适合标记核酸(例如基因组dna)的任何标记方法。
在又另一个实施方案中,避免用可检测标记对样本和参照核酸例如dna单独地进行共价标记。例如,未标记的基因组dna样本与固定至编码颗粒的扩增子杂交。事先标记的报告序列也在与所述扩增子的俘获探针序列相邻但不重叠的序列处与该扩增子-样本dna复合物和扩增子-参照dna复合物杂交。所述经标记的报告序列可以在同一或者不同的杂交反应中被杂交。通过这种方式,所述经标记的报告序列可以在更大规模的环境中被大量生产,与在分析时单独地标记每一个样本相比,降低了每个分析的成本。
“样本”和“参照”核酸例如基因组dna可以获自任何适合的来源。本文描述的具体方法包括使用受试个体的样本基因组dna。基因组样本和/或参照dna可以从几乎任何的组织中提取,所述组织包括但不限于血液、羊水、实体瘤、活检器官、颊部拭子、绒毛膜绒毛、胚泡和卵裂球、妊娠物、唾液、尿液等。从经福尔马林固定、石蜡包埋(ffpe)的病理学样本提取的留存样本同样也是利用此方法分析的样本基因组dna的来源。样本和/或参照基因组dna还可以获自体外来源如细胞系。从这些来源或其他来源获得基因组dna的方法为本领域所熟知。
在具体的实施方案中,就待分析的样本dna的具体特征来对参照dna进行表征。例如,如果打算对样本dna进行分析以检测具体基因或染色体座位的复制情况,则对参照dna进行表征,从而获知有多少个拷贝的该基因或者座位包含在参照dna中。通常,使用来自相同物种的样本和参照dna。
参照dna可以是来源于性别相同的多个正常受试者,特别是人类受试者的基因组dna的汇集混合物。汇集自多个正常受试者的dna可以商购获得。
在一些实施方案中,使用了不止一种参照dna,并在使用其他参照dna的方法中获得其他的信息。
因此,例如,在一个具体的实施方案中,将两种参照基因组dna样本与测试基因组dna样本进行比较。将获自男性受试者的第一种参照基因组dna和获自女性受试者的第二种参照基因组dna与获自性别有待确定的受试者如产前胎儿的测试基因组dna样本进行比较。
本发明分析包括将至少两种参照基因组dna样本与测试基因组dna样本相比较,所述分析的一个具体特征是减少了分析结果的模糊度,并增加了其可信度。例如,如图14所示,在使用男性特异参照和女性特异参照进行分析时,仅用男性特异参照获得的模糊结果被弄清楚。
可以使用本文所述的分析来检测或者表征与染色体获得或丢失相关的障碍。先天的或者天生的障碍包括全部染色体的三体性、较小基因组座位(大约200kb至20mb)的扩增或缺失以及亚端粒区域或着丝粒区域的扩增或缺失。许多癌症的特征也在于可能与类型、阶段、药物抗性或治疗响应有联系的染色体获得和丢失。可以使用本方法就染色体稳定性对实验室细胞系(包括干细胞系)进行表征。
因此,可以使用两种或者更多种参照基因组dna样本,所述样本代表影响到基因组dna的进行性疾病、病症或障碍的不同阶段,并且将所述两种或者更多种参照与一个基因组dna测试样本进行比较,所述测试样本来自欲相对于所述参照来确定其病症的受试个体。例如,将各种癌症、其他障碍和/或年龄与基因组dna的进行性缺失联系起来,例如线粒体dna缺失与端粒变短。
尽管本文所描述的方法和组合物主要涉及来源于人的核酸,但是可以理解的是,也可以将本文所述的方法和组合物用于分析来源自许多生物体中的任一种的分析样本基因组dna,所述生物体包括但不限于非人类的灵长类、啮齿动物、兔、狗、猫、马、牛、猪、山羊和绵羊。还可以分析非哺乳动物来源的样本dna,举例来说,包括鱼和其他水生生物、鸟类、禽类、细菌、病毒、植物、昆虫、爬行类、两栖类、真菌和分支杆菌。同样地,参照dna也可以为人类dna或者来自于各种生物体中的任何一种的dna,所述生物体包括但不限于非人类的灵长类、啮齿类、兔、狗、猫、马、牛、猪、山羊、绵羊以及非哺乳动物来源,举例而言,所述非哺乳动物来源包括鱼和其他水生生物、鸟类、禽类、细菌、病毒、植物、昆虫、爬行类、两栖类、真菌和分歧杆菌。
所述与基底相连的核酸探针与受试个体的可检测地标记的样本基因组dna杂交,从而实现所述与基底相连的核酸探针与所述样本和/或参照核酸的特异性杂交。
在本文描述的分析的具体实施方案中,与编码颗粒相连的扩增子与受试个体的可检测地标记的样本基因组dna杂交,从而实现所述扩增子dna与所述可检测地标记的样本基因组dna的特异性杂交。此外,与编码颗粒相连的dna序列与可检测地标记的参照基因组dna杂交,由此实现所述扩增子dna和所述可检测地标记的参照基因组dna的特异性杂交。
术语“杂交”是指互补核酸的配对和结合。出现在两个核酸之间的杂交程度因诸如核酸的互补程度、核酸的解链温度tm和杂交条件的严格度等因素而异,正如本领域所熟知的那样。术语“杂交条件的严格度”指与具体的常用添加剂如甲酰胺和denhart’s溶液相关的杂交介质的温度、离子浓度和组成条件。与特定的核酸相关的具体杂交条件的确定是常规的,并且为本领域所熟知,例如如j.sambrookandd.w.russell,molecularcloning:alaboratorymanual,coldspringharborlaboratorypress;3rded.,2001;和f.m.ausubel,ed.,shortprotocolsinmolecularbiology,currentprotocols;5thed.,2002中所述。高严格度的杂交条件为仅允许基本互补的核酸进行杂交的那些杂交条件。通常,互补度为约85-100%的核酸被认为是高度互补的,会在高严格度条件下杂交。中严格度条件的实例有互补度居中(互补度为约50-84%)的核酸以及互补度高的核酸进行杂交的条件。相反,低严格度的杂交条件为互补度低的核酸进行杂交的那些杂交条件。术语“特异性杂交”和“特异性地杂交”是指一具体的核酸与样本中的目标核酸进行杂交,但基本不与目标核酸之外的核酸杂交。
所述分析可以在任何合适的容器中进行。在具体的实施方案中,例如,在准备分析多个样本时,可以使用多室容器。举例说来,多室容器包括多坑基底,如载玻片、硅片或硅盘(silicontray)。在一些实施方案中,将每个样本放于多孔板不同的孔之中。例如,多孔板可以是96-孔、384-孔、1024-孔或者1536-孔分析板。
还包括对第一信号的检测和对第二信号的检测,所述第一信号指示所连接dna序列与受试个体的可检测地标记的基因组dna的特异性杂交,所述第二信号指示所连接dna序列与可检测地标记的参照基因组dna的特异性杂交。
使用任何合适的方法来在本文所述的分析中检测信号,举例说来,所述方法包括光谱法、光学法、光化学法、生物化学法、酶学法、电学法和/或免疫化学法。
通过评估一个或者多个可检测标记的信号,可以检测每一颗粒指示杂交程度的信号。通常对各颗粒逐一进行评估。例如,可以使颗粒通过流式细胞仪。示例性流式细胞仪包括beckmancoulter公司(fullertoncalif.)的coulterelite-esp流式细胞仪或facscan.tm.流式细胞仪,以及cytomation,inc.,fortcollins,colo的moflo.tm.流式细胞仪。除了流式细胞术之外,还可以将离心机用作颗粒分离和归类的工具。一个合适的系统为描述于美国专利no.5,926,387中的系统。除了流式细胞术和离心之外,还可以将自由流动电泳装置用作颗粒分离和归类的工具。一个合适的系统为描述于美国专利no.4,310,408中的系统。还可以将颗粒置于表面上,并进行扫描或成像。
在某些实施方案中,检测指示所述与基底相连的核酸序列与可检测地标记的样本核酸(如受试个体的基因组dna)特异性杂交的第一信号。还检测指示所述与基底相连的核酸探针与可检测地标记的参照基因组dna特异性杂交的第二信号。
在其他实施方案中,检测指示所述与编码颗粒相连的dna序列与受试个体的可检测地标记的基因组dna特异性杂交的第一信号。同时,检测指示所述与编码颗粒相连的dna序列与可检测地标记的参照基因组dna特异性杂交的第二信号。
比较所述第一信号和所述第二信号,从而获得有关所述样本和参照核酸的信息。在某些实施方案中,比较所述第一信号和所述第二信号,从而获得有关受试个体的基因组dna与参照基因组dna相比较的信息。
在具体的实施方案中,将与一个或者多个颗粒系列的扩增子杂交的参照dna和样本dna的可检测标记的信号比用来评估该样本和参照dna之间的差异,所述差异指示例如基因组获得和/或丢失。
在某些实施方案中,参照dna和样本dna与一个或者多个颗粒系列的探针例如扩增子在同一个容器(如多孔板的一个孔)中杂交。杂交之后,两个标记在一起分析,即在杂交物中同时检测到两个可检测标记,或者,将杂交物分成两个(或者更多个)部分,分别地对每一个部分进行评估,从而对可检测标记进行检测。可以将评估结果用来提供所述两个可检测标记的信号比。这种方法使得可以使用竞争性杂交来对各分析之间的任何变异进行标准化:在加入有相同颗粒的同一容器中同时分析参照和实验样本。
任选地,可检测地标记的参照dna和可检测地标记的样本dna与一个或多个颗粒系列在不同的容器(例如,多孔板的不同孔)中杂交。在另一个实例中,可检测地标记的参照dna和可检测地标记的样本dna与连接在平面阵列不同位置处的一个或者多个探针杂交。可以获得这两个可检测标记的信号比,以评估所述样本dna和参照dna之间的差异。在使用这种方法时,几个或者多个实验样本可以共用一个参照样本。对于每天涉及多个样本的实验,通过避免对多个双份正常样本进行标记,可以节约试剂和人工成本。同时,也没有必要对样本进行处理,从而获得不同的部分以便分别进行分析。每个样本都可以仅评价一次。
在具体的实施方案中,通过其编码信息来对编码颗粒进行鉴定,从而将颗粒编码情况与第一信号和第二信号联系起来。因此,例如,将第一和第二信号与含有人13号染色体dna的第一编码颗粒系列的编码颗粒关联。对所述与第一编码颗粒系列关联的第一信号和第二信号进行比较,从而获得受试个体13号染色体dna与13号染色体参照dna相比较的信息。类似地,将第一和第二信号与含有人18号染色体dna的第二编码颗粒系列关联。对所述与第二编码颗粒系列关联的第一信号和第二信号进行比较,从而获得受试个体18号染色体dna与18号染色体参照dna相比较的信息。
本文的附图和描述对最佳方式进行了阐释,但是可以用许多备选材料和方法进行替代。本领域的技术人员会认识到合适的备选材料和方法,并且无需进行过多试验即可制备和使用所述的组合物和方法。
多种缓冲液和其他分析组分的组成均可被替代。
用于培养、纯化、扩增、变性、偶联、杂交、报告物连接、洗涤和小球处理的条件全都可以由使用者来改变,从而适合具体类型的细胞、模板基因组dna、样本、所选报告物等。
使用少到30ng的样本dna也可以很好地进行本文实施例部分的分析。在生物学来源产生的用于所述分析的dna不足的情况下,可以通过许多全基因组扩增(wga)方法(如doppcr或phi-29pcr)对样本进行扩增。使用wga处理的样本时,可以以同样的方法对参照dna进行处理,从而使任何序列特异性扩增的偏差均可以基本由样本/参照的信号比来校正。
提供了用于分析dna的试剂盒。在具体的实施方案中,提供了一种试剂盒,其含有编码颗粒系列和/或两个或更多个编码颗粒系列的混合物。任选地,试剂盒含有使用编码颗粒系列和/或含有两个或者更多个编码颗粒系列的多重试剂的说明材料。任选地,还含有辅助试剂,如缓冲液、酶、洗涤溶液、杂交溶液、可检测标记、检测试剂等。
下文的实施例对用于分析的组合物和方法的实施方案进行了阐释。提供这些实施例的目的是进行说明,不能将其看作是对组合物和方法的范围进行限制。
实施例
实施例1
用于基因组dna分析的小球系列试剂的制备
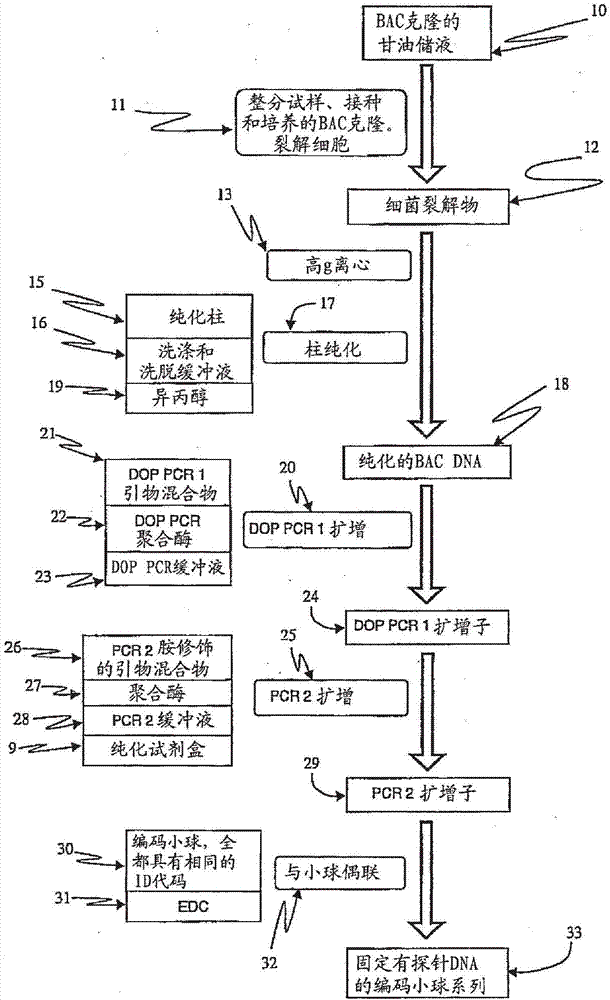
图1a示出了由单个bac克隆制备bac扩增子并将该扩增子作为探针固定到一个系列的编码小球上的流程图。在此实施例中,该系列中的小球全都具有相同的id代码。
原料为活的bac克隆材料(10),即一段插入到大肠杆菌细菌细胞基因组内的长(通常为100-200kb)的人dna序列。取出一小片冷冻的bac甘油储液材料,并将其用作标准细菌细胞培养方法的原料(11)。根据标准的bac培养方案,在37℃下于装有35ml培养基的50ml管中过夜培养细胞,并用抗生素进行选择。然后在4℃将培养得到的细胞离心20分钟,使其离心到管底,取出并弃去上清液。将细胞沉淀重悬于含有rnase的缓冲液中,然后使用lyseblue(qiagen,valenciaca)和sds裂解。以大约20,000g将裂解液(12)离心(13)30分钟,并收集上清(溶液中含有dna),弃去沉淀。将上清液重复离心15分钟。收集含有溶解的bacdna的澄清上清液,同时弃去离心到管底的细胞碎片、蛋白质和其他杂质。使用qiagen基因组-tip20/g柱纯化试剂盒,从上清液中提取和提纯bacdna(17)。此试剂盒含有纯化柱(15),以及洗涤和洗脱缓冲液(16)。洗脱之后,通过异丙醇(19)沉淀法将现已高度纯化的bacdna进行沉淀并成为沉淀团块。产量通常是20至200ng的纯化bacdna(18)。可以将此bacdna以干燥沉淀物形式储存,或者重悬于水中,直接用于随后的步骤中。
然后,使用两轮聚合酶链式反应(pcr)扩增,制备基本上代表各bacdna的完整序列物的大量pcr扩增子。第一轮pcr(20)是非特异性简并寡核苷酸引物(dop)的pcr,其使用dop引物混合物(21)、doppcr聚合酶(22)和doppcr缓冲液(23),并将上面制备的bacdna(18)用作模板。第二轮pcr扩增(25),利用针对dop引物的已知序列基序的单个引物。使用两轮pcr来产生产量为大约20μg的扩增子终产物(29),用于随后将扩增子(29)偶联(32)至编码小球(30)。
如下制备扩增子。
制备50μl各bacdna的第一doppcr混合物,其含有:
doppcr缓冲液(23)含有20mmtrishcl(ph8.4)、50mmkcl和5mmmgcl。dntp(amershambiosciences,piscatawaynj)的浓度为200μm。platinumtaq聚合酶(appliedbiosystems)的浓度为5单位/μl。dop引物混合物(21),参见fiegleretal.2003,geneschromosomescancer,36(4):361-74,包括三个系列具有以下22-mer序列的简并寡核苷酸(operonbiotechnologies,huntsvilleal),其中n代表随机的核苷酸:
5'ccgactcgagnnnnnnctagaa3'seqidno.1
5'ccgactcgagnnnnnntaggag3'seqidno.2
5'ccgactcgagnnnnnnttctag3'seqidno.3
其中n表示随机的核苷酸。
使用qiagen基因组-tip20/g柱纯化试剂盒,将溶于水的bacdna模板(18)通过柱纯化法(17)纯化。platinumtaq聚合酶(22)(invitrogen,carlsbadca)的浓度为5单位/μl。
根据以下温度/时间条件,在geneamp9700热循环仪(appliedbiosystems,fostercityca)中进行第一轮扩增(20):
3.0分钟94℃
1.5分钟94℃
2.5分钟30℃,9个循环
0.10c/秒72℃(斜率)
3.0分钟72℃
1.0分钟94℃
1.5分钟62℃,30个循环
2.0分钟72℃
8.0分钟72℃
4.0℃(稳定状态)
然后,将第一轮doppcr(20)的扩增子产物(24)用作第二轮pcr(25)的模板。第二轮中的单一引物(26)具有针对第一轮(20)中使用的dop引物的共有序列部分的特异性。此引物(26)经胺修饰,从而使所得扩增子(29)还在一个末端具有胺基团,以有助于在下一步中简单地偶联至编码小球(32)。
如下进行第二轮pcr。
制备100μl各bac扩增子模板的第二pcr混合物,其含有:
pcr2缓冲液(28)含有20mmtrishcl(ph8.4)、50mmkcl和5mmmgcl。dntp(amershambiosciences,piscatawaynj)的浓度为200μm。platinumtaq聚合酶(appliedbiosystems)的浓度为5单位/μl。
与胺相连的引物(operon)具有以下序列。
5'-ggaaacagcccgactcgag-3'seqidno.4
反应(25)中的模板为来自上一轮doppcr(20)的dop扩增子。根据以下温度/时间条件,在geneamp9700热循环仪(appliedbiosystems)上进行第二轮扩增(25):
10分钟95℃
1.0分钟95℃
1.5分钟60℃,35个循环
7.0分钟72℃
10分钟72℃
4.0℃(稳定状态)
然后,使用基于磁性小球的试剂盒(9)(pcrcleanbeads,agencourtbiosciencecorp.,beverleyma),根据制造商的方案,纯化此第二pcr产物(29)。随后将经纯化的扩增子(29),重悬于40μl水中,并于-20℃下储存,直至用于下文所述的小球偶联步骤中。
以50μl的标准小球浓度的规模,在luminex羧基小球(30)(luminex,austintx),上进行编码小球偶联过程(32),以将所述扩增子产物(29)作为探针dna固定到编码小球的表面之上,得到大约650,000个小球。所述小球由聚苯乙烯制备,直径大约为5.6μm,并由受控量的两种或者多种荧光染料编码,从而有助于在专用流式细胞仪读取装置上检测其小球id。将50μl的悬浮小球(30)(全都具有一个小球id或区域),从送递它们的luminex管中转移到一个1.5mleppendorf管进行偶联(32),并通过涡旋和超声处理来确保悬浮。然后,以12,000rpm将小球离心3分钟,并在不干扰小球沉淀物的情况下除去小球缓冲上清液。将25μlmes缓冲液加入各小球管,然后进行涡旋和超声处理。另外地,将10μg各bac的pcr2扩增子(29)加入第二系列的1.5ml离心管中,随后将各管中的dna在speedvac(thermofisherscientific,walthamma)中彻底干燥。然后,将一份小球悬液转移到各dna管中,并将各管涡旋和超声处理5秒钟进行混合,仔细地跟踪与各bac有联系的小球id(区域)。
接下来,将1.5μl刚溶解的edc(31)(1-乙基-3-[二甲基氨丙基]-碳二亚胺盐酸盐,pierce,rockfordil)以10mg/ml的浓度加入各管中,立即涡旋,并在室温下避光(目的是保留luminex小球的荧光编码)孵育30分钟。在15分钟时再次混合。然后再一次地重复edc添加、孵育和再次混合。
然后,将500μltnt缓冲液(0.1mtrisph7.5,0.15mnacl,0.02%tween20)加入各管中并进行涡旋。然后在微型离心机上以12,000rpm将所述管离心4分钟,使小球分布到底部,并小心地除去上清液。接下来,加入500μl0.1%sds,并且再以12,000rpm将小球离心4分钟,小心地除去上清液。最后,将50μl1×te缓冲液(10mmtrisph7.5,1mmedta)加至各管,并进行涡旋。
该小球系列(33)——固定有扩增子探针(29)——可以作为用于基因组dna分析的多重小球系列的一个组分纳入。
实施例2
dna分析用多重编码小球系列试剂的制备
图2是示出将m个不同的编码小球系列(各自具有各自的固定的bac-扩增子探针dna)混合在一起来制备多重编码小球系列的流程图。
通过超声处理、旋转管容器、涡旋或类似方法,将编码小球系列34、35、36和37制成悬液。然后,使用移液管将各小球系列的整分试样转移到另一个容器中,各个小球系列在该容器中合并和混合,然后变性(38),从而有助于随后在分析中与固定在所述小球上的探针dna的杂交。
在一个详尽的实施例中,将各50μl的2个或多个小球系列——每一系列在一个单独的管子里,每一编码小球系列均固定有探针dna(33)——分批地合并到一个1.5ml离心管内。在合并大约10个小球系列之后,将管离心,并小心除去上清液,目的是减少体积。再次重复,直至合并所有的小球系列(例如,luminex200系统至多支持100个编码小球id或区域)。
在将所有的小球系列合并成为一个多重小球系列之后,将固定的探针dna变性。在对小球进行离心并除去上清之后,加入500μl0.1nnaoh,并在室温下孵育2分钟。然后对小球进行离心,并小心地除去上清。加入500μl10mmtris、15mmnacl、0.2%tween20,涡旋该管,然后对小球进行离心,并除去上清液。然后重复进行此洗涤步骤。最后,用1×te缓冲液将体积调整为500μl,并将此多重小球系列(39)在4℃下避光保存,直至用于分析中。
实施例3
多重基因组获得和丢失分析
图3是展示一个实施方案的流程图,所述实施方案包括使用多重编码小球系列在n个样本上进行多重基因组获得和丢失分析。该流程图示出了方法的实施方案,包括提供经标记的样本和参照dna(5),所述样本和参照dna与两个或者更多个编码小球系列杂交(6),检测所述标记样本和参照dna与所述编码小球系列杂交的信号(7),和比较所述信号以确定所述样本和参照dna之间的差异(8)。
图3a为展示一个实施方案的流程图,所述实施方案包括:使用多重编码小球系列对n个样本进行多重基因组获得和丢失分析。
在此实施例中,平行分析了两个dna样本和两个参照。事实上,可以同时地以微量板形式平行分析几十个样本。也可以平行分析比此数目更多或者更少的样本和参照。
在此实施例中,将四个dna样本(40和41代表两个参照,而42和43代表两个分析样本)用生物素进行酶学标记并纯化。参照样本通常为正常男性和女性的合并样本,如人类女性基因组dna和人类男性基因组dna(promega,madisonwi)。将各dna样本和参照与生物素标记的核苷酸(45)(perkinelmer,bostonma)、非标记核苷酸(49)(perkinelmer)、随机引物(47)(operon,biotechnologies,huntsvilleal)和klenow片段聚合酶(46)(epicentrebiotechnologies,madisonwi)合并起来。孵育(44)之后,使用dna柱纯化试剂盒(49)(如purelinkdnaminikit(invitrogen))纯化(50)反应产物。大约5μl的约200ng/μl经标记的样本用于此分析随后的杂交。
然后,使各生物素标记的样本或参照(51-54)与固定在多重编码小球系列(56)的小球上的探针杂交(55)使用了每个小球系列(各探针类型)的大约500个小球;在此55重实施例中,使用了总共大约55×500=27,500个小球/杂交。
因为编码情况,各编码小球系列的小球可以区别于其他编码小球系列每一系列的小球。此55个小球系列中的每一系列均含有多个编码小球,与所述编码小球相连的扩增子基本上代表完整的模板基因组dna片段。各小球系列的模板dna代表图9所列的一个基因组座位。
杂交反应中包括含有cot-1dna、甲酰胺、硫酸葡聚糖和1.9×ssc的杂交缓冲液。总体积大约为15μl,且反应在硬的pcr型硬质微量板如bio-radhsp9631(bio-radlaboratories,herculesca)的孔中进行。使用铝箔封口机(msf1001,bio-rad)严密地封住该板,将蒸发降至最低。在1150rpm的微量板振荡培养箱(wallacncsincubator,perkinelmer)中于50℃过夜进行杂交孵育(55)。
在杂交孵育(55)之后,与四个样本杂交的四个多重小球系列(58-61),就可以进行杂交洗涤(63),然后与荧光报告物(65)一起孵育,再进行报告物洗涤(67)。首先,将100μl洗涤缓冲液a(2×ssc,50%甲酰胺)加入各孔之中,然后重新封闭该板,并在振荡培养箱(转动速率为1150rpm)于50℃下孵育20分钟。然后将各孔内含物转移到millipore0.46μmht过滤板(millipore,billericama)。之后,使用milliporemsvmhtsoo多头抽真空装置从各孔中真空除去液体。接下来,将100μl洗涤缓冲液b(2×ssc,0.1%igepal清洁剂)加入各孔,然后在50℃下再进行一次为期20分钟的振荡孵育,并进行真空抽吸。随后,将100μl洗涤缓冲液c(0.2×ssc)加入各孔,并在50℃下重复为期20分钟的振荡孵育,然后进行真空抽吸。
然后,将100μl1×phycolinksa溶液(链亲和素-藻红蛋白报告物),64,加入各孔。此报告物溶液是通过将2μl500×phycolinksapj13s(prozyme,sanleandroca)混合到1ml报告物稀释液中形成,其中所述稀释液为1×pbs、0.1%bsa和0.05%tween20。将此报告物溶液与多重小球系列在1050rpm的振荡培养箱中于25℃下孵育30分钟。孵育之后,使用如前面的洗涤步骤中的多头抽真空装置从过滤板的孔中吸出溶液。
然后用洗涤缓冲液d(66)将小球洗涤两次(67),所述洗涤缓冲液d为含0.01%tween20的1×pbs。将100μl加入过滤板的各孔中,然后通过板孔底部的过滤器真空抽吸掉液体。再一次加入100μl,并在1050rpm的振荡培养箱中于25℃下孵育2分钟。所述第二次洗涤并不进行抽吸,而是用来悬浮小球以进行读数。
之后,本实施例中的四个小球系列(68-71)即可在luminex200系统(luminexcorporation,austintx)上读数(72)。依次读取各孔中的小球的信号和小球id,并记录下各孔或样本的各小球id(小球区域)的前50个小球的荧光强度中值,并输出到在一个数据文件(73)中。没有发现小球交联;设定luminex读数仪,使其分析各区域的50个小球,并且并未记录到失败。
图4是96孔sbs标准微量板(80)的示意图,其示出用于平行地对46个样本进行分析的两份参照和两份样本的示例性位置。各标记样本的两份重复杂交可用于确保孔密封失败情况下的数据形成,所述孔密封失败会导致试剂从单个孔蒸发。在双份重复没有受到影响时,该样本仍然形成数据。使用此微量板和编码小球方法,实验员单人即可同时分析例如46个样本和2个参照,并且全都双份重复进行,在第一天进行标记,杂交过夜,并在第二天进行洗涤和读数。或者,分析可以不重复进行或者不止两个重复地进行。所示出的是双份的两个参照(81和82)以及双份的样本,样本的一个实例在83处标示出。
图5是使用男性的13号染色体上具有三体性的corielldna样本形成数据的一个实例;
此数据由通过luminex读数仪获得的各小球区域的中间荧光值计算得到。从所有其他信号中减去阴性对照小球29、54和56的平均值(参见图9)。然后,求出来自九个常染色体克隆的信号与来自男性和女性参照dna的相应克隆信号的比值。计算标准化因子,从而在将该因子应用到所有的常染色体克隆信号之时,其会使得平均的常染色体比值为数值1。然后,将此标准化因子应用到所有的样本信号。
将所得比值作图,并示于图5。请注意,13号染色体的克隆的比值全都在范围1.3至1.6之间,而18和21号染色体的克隆以及其他常染色体克隆除了有一个之外全都低于1.2。13号染色体中的三体性很容易显现。同样地,样本与男性参照相比的比值图(方块数据点)对于x和y性染色体实际上都是平坦的。此乃男性样本所应有的反应。样本与女性参照相比较的图(菱形数据点),对于x而言下移,而对于y而言上升,这同样也是男性样本应有的反应。
图6是使用男性的18号染色体上具有三体性的corielldna样本所形成的数据的一个实例。如针对图5所述的那样,形成数据并作图。
图7是使用女性的21号染色体上具有三体性的corielldna样本所形成的数据的一个实例。如针对图5所述的那样,形成数据并作图。
图8是使用x染色体5-拷贝扩增的corielldna样本所形成的数据的一个实例。如针对图5所述的那样,形成数据并作图。
图9为一张图表,其示出具有用于在示例性分析中形成扩增子的人基因组dna插入物的bac克隆、其染色体和cyto条带位置、阴性对照寡核苷酸的序列以及各扩增子探针所固定的小球系列的小球id(luminex小球区域)。将图5-8中x轴上顺序编号的作图点与图9中自上而下列出的bac关联。bacrp11-186j16被固定到两个不同的小球区域(42和86)。
与人基因组没有序列同源性的一个寡核苷酸被选择作为阴性对照。所使用的具体阴性对照寡核苷酸为
5'gtcacatgcgatggatcgagctc3'seqidno.5;
5'ctttatcatcgttcccaccttaat3'seqidno.6;
5'gcacggacgaggccggtatgtt3'seqidno.7。
将与阴性对照寡核苷酸相连的三个小球区域29、54和56所形成的信号取平均值,并从所有其他小球信号中减去,然后再计算比值。
实施例4
图10a是示出根据本文所述一个方面的一种制备复合探针的方法的简化流程图。来自于一个来源的探针dna(92)和来自于另一个来源的探针dna(93)任选地通过pcr分别被扩增(94和95),从而得到扩增子探针,随后混合所述扩增子探针形成复合探针(96)。将所述复合探针连接至基底(97)形成与基底相连的复合探针(98)。
图10b是示出根据本文所述一个方面的一种制备复合探针的方法的简化流程图,其中可以将探针dna(92和93)汇集形成复合混合物99,然后进行任选的pcr扩增(100),从而制备复合探针材料(96),所述复合探针材料连接至基底(97)形成与基底相连的复合探针(98)。
图11是示出根据本文所述方法一个方面的一种制备复合探针的方法的简化流程图。示出了一幅染色体模式图(101),该模式图示出所关注的染色体(在本实施方案中,是22号染色体)的cytoband(102)。示出用于分析的具有目的区域中基因组座位的一个五种bac的系列(103),且各bac基因组座位大致被搁置于所述染色体模式图上。在本实施方案中,来自五种bac且被定位到与digeorge微缺失综合征相对应的cytoband22p11.2的dna被用于制备所述复合探针。该图中的所述染色体模式图示意性地示出选自一个cytoband的五种示例性bac的基因组邻近关系;在此方法中,所述bacdna并非从人染色体提取。
利用常规方案从所述五种培养的bac中的每一种中提取和纯化dna。将所培养的细菌细胞裂解,并通过离心和柱纯化(qiagen,valenciaca)来沉淀并随后纯化dna(104)。随后,将从各bac中纯化的dna(105)用作简并寡核苷酸引物(dop)pcr扩增(106)的模板。接下来,将dop引物序列用作特异性pcr引物,对所述dop产物进行特异性pcr扩增。这一方法得到了五种单独的扩增子探针(107)。接下来,将这些单独的探针(107)汇集起来(108),形成复合探针(109)。然后,将所述复合探针固定到一个系列的luminex编码多重微球(全都具有一个小球“区域”,即具有相同的小球编码身份)(110),用于在多重基因组获得-丢失分析中用作22p11.2cytoband探针。所述单独的探针(107)的每一种也被单独地固定,每一种被固定到具有一个独特身份的小球系列上,从而使得其反应可以与所述复合探针的反应相比较。
图12是测试分析的数据曲线图,示出了复合探针与digeorge综合征参照dna样本(coriellinstituteformedicalresearch,camdennj)的使用。使用固定在luminex编码微球上的pcr产物探针在luminexxmap平台上进行分析。使用doppcr由bacdna制备所述pcr产物探针。固定所述探针,微球系列上的每一种探针都可以被luminex系统分别鉴定,并如本文所述进行测试分析。所述多重探针组包括八种来自基因组座位的常染色体探针,所述常染色体探针预计在dna样本与男性和女性dna参照之间没有表现出获得或者丢失。所述多重探针组还包括六种x染色体探针和五种y染色体探针作为阳性对照,从而在将测试样本与相反性别的参照进行比较时,可以观察到性染色体中已知的获得或者丢失的比值反应。所述多重探针组还包括位于digeorge综合征缺失的座位处的五种22q11.2探针(123),参见下表i。所述五种bac的中心座位跨越大约0.45mb(445kb),并且对于其175kb的典型长度,总跨度略高于600kb。
表i.bac及其中心的chr22线性定位位置(mb)
参照图12,此为digeorge综合征样本与男性和女性正常参照dna两者相比较的两个数据系列的比值图。数据系列120是digeorge样本与男性参照dna相比较的比值反应;其表明所有x染色体探针中均有相对的获得(125),而在所有y染色体探针中均有丢失(126)。该反应表明,所述样本来自于女性。数据系列121是同一样本与女性参照dna的比较,其表明,对于x和y性染色体探针,比值反应均接近1.0,这确认了该样本来自于女性。图中1.0的比值线(122)指示在样本与正常参照相比没有基因组获得或者丢失时,对于任何给定探针而言,样本/参照比值的预期结果。将常染色体探针(127)加入到此组,作为预期会产生大约1.0比值的对照,它们的确如此。
来自于22q.11.2座位的五种探针(123)均包含在此组中。这五种探针与男性和女性参照dna相比,全都表现出比值小于1,这与所述样本中在该座位处的已知基因组缺失相一致。最后,将一个类型的多重小球偶联到含有所述五种22q11.2探针的混合物或汇集物的复合探针(123)。所述复合探针的比值反应(124)也指示缺失,其与所汇集的所述五种组成型探针的平均反应相当一致。
实施例5
图13是根据本发明的一个方面由luminex小球阵列获得-丢失分析得到的比值数据。该数据来自于从产前羊水样本提取的胎儿dna,所述胎儿的性别并不明确知晓。进行分析,并且男性和女性参照在同一96-孔微量板的不同孔中同时进行分析。图例(136)标识出两个数据标记,表示参比女性的(菱形作图点)和参比男性的(方形作图点)。比值=1.0的水平线(132)位于该图的中心。所述比值标尺(133)为该图的纵轴。示出了具有针对女性参照形成的样本/参照比值的第一数据图(130)。对于该图,针对x染色体探针的数据(134)表现出比值小于1,针对y染色体探针的数据(135)与女性相比表现出获得。这与男性样本是一致的。第二数据图(131)利用接近比值等于1.0的线的男性参照群,也与男性样本一致。两个数据系列都显示出阵列中的其他探针并没有明显地偏向x探针的左侧,表明是一个正常样本。
表ii示出与图13和14中x轴上顺序编号的作图点有关的bac身份。
表ii.
auto表示常染色体。
图14为使用具有先前表征的基因组异常(三倍体18和xxx)的样本(coriellinstituteofmedicalresearch,trentonnj),来自同一luminex小球阵列分析获得的比值数据。同样地,该样本参比女性(145)和参比男性(146)的两个数据图被同时展示。三倍体18的探针,全都产生与两个参照相比表现出获得(142)的比值数据。在参比女性的图(145)上,x探针的数据显示出获得(144)。此获得的幅度与三倍体获得(142)的幅度相同,这与xxx(样本)/xx(女性参照)比值的结果一致。参比男性的图(146)在x探针上显示出大得多的获得(143),正如以xxx(样本)/x(男性参照)比值所预期的那样。针对y染色体的探针有噪音,这在acgh中很常见,但是其他的探针全都紧紧地簇拥于比值等于1.0的线(141)周围。比值标尺(141)是该图的纵轴。
这些实施例表明,根据针对男性和女性参照形成的比值数据,具有正常的x和y染色体组分的样本的性别是显而易见的。对于多x异常样本同样显而易见的是,对多拷贝的x的定量在使用两个参照时更为直接。
本说明书中提及的任何专利或出版物均通过援引纳入本文,其程度就如同每一份出版物都逐一特别地被指出通过援引纳入。以下申请均通过援引全文纳入本申请:2006年12月22日提交的流水号为11/615,739的美国专利申请;2008年3月26日提交的流水号为12/055,919的美国专利申请;以及2005年12月23日提交的流水号为60/753,584的美国临时专利申请、2005年12月23日提交的流水号为60/753,822的美国临时专利申请、2006年2月3日提交的流水号为60/765,311的美国临时专利申请、2006年2月3日提交的流水号为60/765,355的美国临时专利申请和2007年12月5日提交的流水号为60/992,489的美国临时专利申请。
本文描述的组合物和方法以示例的方式代表本发明的某些实施方案,而并无意于限制本发明的范围。本领域技术人员可以想到其改变以及其他用途。在不背离本发明权利要求所列出的范围的情况下,可以作出所述变化和其他用途。
序列表
<110>珀金埃尔默.拉斯公司
<120>与多重基因组获得和丢失分析相关的方法和组合物
<130>nen-23603/16
<150>11/615,739
<151>2006-12-22
<150>60/992,489
<151>2007-12-05
<150>12/055,919
<151>2008-03-26
<150>60/765,355
<151>2006-02-03
<150>60/765,311
<151>2006-02-03
<150>60/753,822
<151>2005-12-23
<150>60/753,584
<151>2005-12-23
<160>7
<170>patentinversion3.5
<210>1
<211>22
<212>dna
<213>人工序列
<220>
<223>简并寡核苷酸
<220>
<221>misc_feature
<222>(11)..(16)
<223>n为a、c、g或t
<400>1
ccgactcgagnnnnnnctagaa22
<210>2
<211>22
<212>dna
<213>人工序列
<220>
<223>简并寡核苷酸
<220>
<221>misc_feature
<222>(11)..(16)
<223>n为a、c、g或t
<400>2
ccgactcgagnnnnnntaggag22
<210>3
<211>22
<212>dna
<213>人工序列
<220>
<223>简并寡核苷酸
<220>
<221>misc_feature
<222>(11)..(16)
<223>n为a、c、g或t
<400>3
ccgactcgagnnnnnnttctag22
<210>4
<211>19
<212>dna
<213>人工序列
<220>
<223>引物
<400>4
ggaaacagcccgactcgag19
<210>5
<211>23
<212>dna
<213>人工序列
<220>
<223>对照寡核苷酸
<400>5
gtcacatgcgatggatcgagctc23
<210>6
<211>24
<212>dna
<213>人工序列
<220>
<223>对照寡核苷酸
<400>6
ctttatcatcgttcccaccttaat24
<210>7
<211>22
<212>dna
<213>人工序列
<220>
<223>对照寡核苷酸
<400>7
gcacggacgaggccggtatgtt22
- 还没有人留言评论。精彩留言会获得点赞!
- 一种新型的15种人乳头瘤病毒亚型分型试剂盒和方法与流程
- 一种与水稻稻瘟病抗性基因紧密连锁的分子标记、引物及其应用的制造方法与工艺
- 一种无创高通量甲基化结肠癌诊断、研究和治疗方法与流程
- 用于检测CDKN2B基因变异的试剂盒及CDKN2B基因变异率的定量检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种癌症相关基因突变高灵敏度检测方法和试剂盒与流程
- 基于高通量测序法的人类多基因突变检测试剂盒的制造方法与工艺
- 一种用于检测德尔卑沙门氏菌的靶基因、特异性引物对及检测方法和试剂盒与流程
- 一种检测测序平台索引序列污染的方法及引物与流程
- 无物种限制的真核生物同时进行基因敲除和基因过表达的制造方法与工艺