一种动物角类药材角质层的DNA提取方法与流程
本发明属于中药材鉴定
技术领域:
,具体涉及一种动物角类药材角质层的dna提取方法。
背景技术:
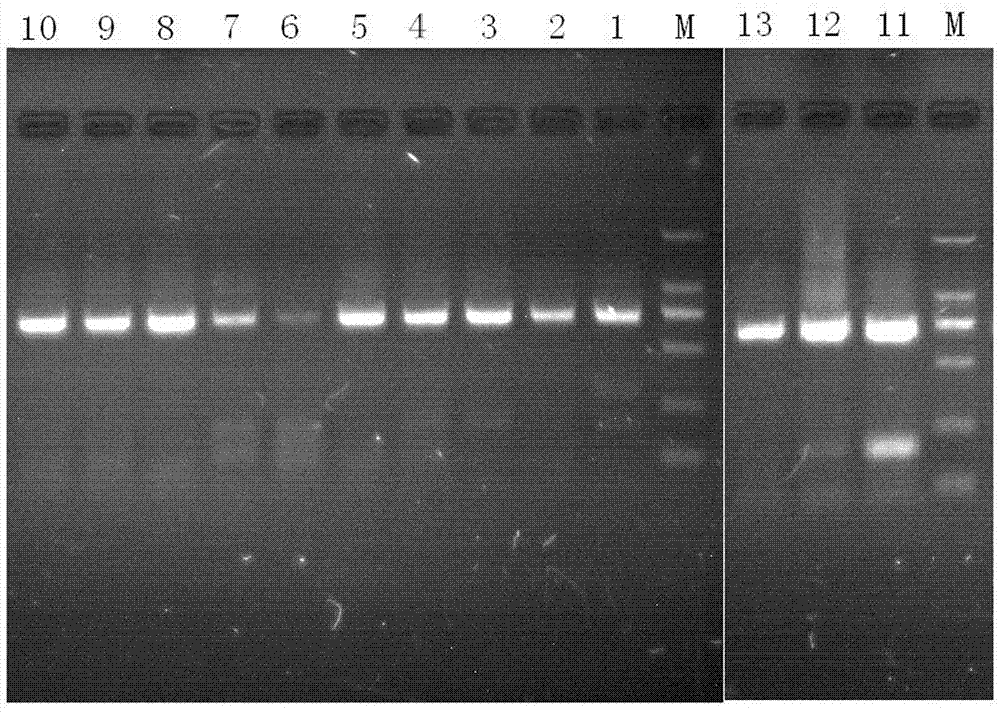
:在祖国医学中,一些动物角有显著的临床疗效。如羚羊角具有平肝熄风,清肝明目,散血解毒的功效;水牛角具有清热凉血,解毒,定惊的功效。因角类动物药材资源有限,价格昂贵,易混品较多,传统鉴定方法主要依赖经验丰富的鉴定专家,一般中药材质量验收人员缺乏性状鉴别经验,难以确保用药有效性。现代分子生物学技术不受样品状态影响,可通过分析其遗传物质dna的差异实现对动物物种的鉴定。存在于动物线粒体的co1基因已在鱼类、鸟类等分类中得到成功的运用,能有效识别新物种,被认为是全球动物物种鉴定的通用条形码,其片段序列可作为种内多样性和种间特异性的一种新的身份识别系统。目前,对于动物药及混伪品的鉴定多通过聚合酶链反应(pcr)技术扩增线粒体上的co1基因,通过序列比对、计算遗传距离、构建系统发育树等来分析正品及混伪品。该技术已用在多种动物药材的不同组织样本如(肌肉、血液)的dna扩增,并取得较好的研究结果。然而,对于角类部位的dna提取,已有文献采用通用动物组织(肌肉、血液)的dna提取方法,该方法对于像角类药材这样的坚硬样本存在角质细胞裂解不彻底,提取出的dna模板浓度低,尤其是长时间放置,dna降解严重的角类药材无法进一步pcr。技术实现要素:本发明为了解决现有动物角类药材dna的提取方法存在角质细胞裂解不彻底,提取的dna模板浓度低,dna降解严重的角类药材无法进行pcr的问题,提供了一种动物角类药材角质层的dna提取方法。本发明由如下技术方案实现的:一种动物角类药材角质层的dna提取方法,包括样品前处理、取样、dna提取,具体步骤为:(1)样品前处理:取收集到的动物角类药材样品,所述动物角类药材样品为个子样品,其处理方法为:先将样品用电锯切割横截面,将样品切割成片状,然后将片状样品用刀片刮去外层污染,取内层,削成细薄片;所述动物角类药材样品为块状样品,其处理方法为:用刀片刮去外层污染后,取内层,削成细薄片;所述动物角类药材样品为丝状样品,其处理方法为:用75%乙醇清洗3次,双蒸水冲洗至无醇味,紫外光下照射,晾干;消毒剪剪成碎片,备用;(2)取样:称取样品25-70mg,加入dtt20-100µl,proteinasek40µl及裂解缓冲液ga200µl后,56℃金属浴酶解3-4小时后提取dna;(3)dna提取:按试剂盒提取步骤提取dna,nanodrop2000测定dna的浓度及纯度;(4)提取的dna样本进行pcr扩增:扩增引物为co1基因的通用引物,正向引物lco14905′-ggtcaacaaatcataaagatattgg-3′反向引物hco21985′-taaacttcagggtgaccaaaaaatca-3′,25µl反应体系,根据nanodrop2000测定dna的浓度,保证模板用量为150~180ng,低于150~180ng,模板取样定为10µl;浓度为10mmol/l的正向、反向引物各1µl;2×easytagsupermix12.5µl;双蒸水补足至25µl;pcr扩增程序为:94℃1min;94℃,1min,45℃,1.5min,72℃,1.5min,5个循环;94℃,1min,50℃,1.5min,72℃,1min,35个循环;72℃,5min;用含有goldviewi型核酸染色剂的1.5%琼脂糖凝胶点样,dl2,000dnamarker为对照,样品5µl与2µl6×loadingbuffer混合均匀点样,tbe缓冲液中电泳,凝胶成像仪成像;割取750bp处的dna条带,胶回收纯化pcr产物,测序;(5)低浓度样品进行二次扩增:样品浓度为10~14ng/µl,过低不能测序时,进行二次扩增:提取的dna第一次扩增后,紫外灯下割取750bp处的条带,切成碎块,100µl双蒸水溶解,60℃温育,模板用量5µl,进行二次扩增;浓度为10mmol/l的正向、反向引物各1µl;2×easytagsupermix12.5µl;双蒸水补足至25µl;扩增程序为95℃1min,40℃,1min,72℃,1.5min,72℃,1.5min,35个循环模,72℃,保存7min;1.5%琼脂糖点样,tbe缓冲液电泳,凝胶成像仪成像,割取750bp处的dna条带,胶回收纯化pcr产物,测序。优选,步骤(2)中称取样品量为25mg,加入dtt量为20µl。采用本发明所述方法对动物角类药材的角质层部位提取动物角中的dna,对于dna严重降解的样本,通过减少溶解dna的溶剂用量,提高初始浓度,或切胶纯化,进行二次扩增,最终使所有样品的pcr反应结果满足测序要求,成功测出所有样品的线粒体co1基因序列。在取材上,样品由于坚硬,手工用刀片刮粉、钢锉锉粉或电动研磨器磨粉,均存在效率低下的缺点,难以完成取样,后改为电锯切割横截面,完成由样品到片状样品取材,片状样品进一步用刀片刮去外层,以防止外原微生物污染。建立角类动物药材的dna提取方法,使提取的dna质量满足常规pcr要求。建立的dna提取方法,提取dna完全,可标准化操作,可应用于多种动物角的提取。采用本发明所述方法可提取角类样品角质层的dna,有效改善常规试剂盒对于角质层样品裂解不透彻,常规方法即使延长时间过夜裂解,也无法改善裂解效果,且提取出的dna浓度低,无法进一步pcr和测序;本发明方法在确定取样量的基础上,加入适当量的dtt,短时间内(4小时内),可使角质层样品完全裂解,保证了dna最大程度提出,也能满足陈旧样品的dna提取,无需加大取样量,无需进口痕量dna提取试剂盒,可满足新鲜和陈旧的动物角类样品,同时避免取骨塞部位(牛角类和羊角类样品含有骨塞)时,用edta脱钙预处理,因脱钙次数和时间不好掌握,造成dna的提取不完全或dna被破坏。本发明方法对于传统鉴定方法无法确定市场上种类繁多的动物角类,通过考察提取过程关键因素,如裂解透彻与否的关键因素dtt、低浓度样品二次扩增时浓度梯度的考察,样品取样量、dtt的加入量等关键因素,保证了所有样品dna最大程度提取及成功进行pcr反应,通过测序、比对确定了动物角的种类,解决了传统方法无法鉴定的不完整样品,保证作为中药材使用的羚羊角、水牛角的正品来源,以防山羊角、黄羊角等羊角类充当羚羊角使用,防止牦牛角、黄牛角等牛角类充当水牛角使用。附图说明图1为收集的10种样品原始照片图;图2为所有样品的pcr图;图3为低浓度样品二次扩增pcr图,图中:1为绵羊角,pcr模板用量为5µl;2为山羊角,pcr模板用量为5µl;3为绵羊角,pcr模板用量为10µl;4为山羊角,pcr模板用量为10µl;5为绵羊角,pcr模板用量为5µl;6为山羊角,pcr模板用量为5µl;图4、图5为所有序列的测序结果图;图6为本发明实验例中所述的陈旧样品分别采用常规试剂盒方法与本发明所述方法进行pcr的对照图,图中:1、2、3为采用常规试剂盒方法进行pcr结果,其中样品分别为羚羊角个子15年、羚羊角片13年、藏羚羊角个子12年;4、5、6为采用本发明所述方法进行pcr结果,其中样品分别为羚羊角个子15年、羚羊角片13年、藏羚羊角个子12年,m为dnamark2000。具体实施方式实施例1:收集河北安国药材市场和安徽亳州药材市场所有角类动物药材及民间收藏的样品(包括羊角类、牛角类),研究角类药材(角质层)的dna提取方法,使提取出的dna质量可满足常规pcr反应要求。1.材料与仪器收集的样品信息见表1。样品照片图见图1。仅凭借图1照片,采用传统鉴定方法无法鉴定所述样品的物种来源信息,而对于dna模板量较小的样品,采用常规dna提取,难以满足pcr测序的要求,因此也无法鉴别出样品的物种来源,凭借主观观察易于混淆样品物种,而发生误判、错判,最终导致用药错误。表1:收集的样品信息血液/细胞/组织基因组dna提取试剂盒(离心柱型),目录号:dp304,天根生化科技有限公司;dl2,000dnamarker,6×loadingbuffer宝生物工程(大连)有限公司;2×easytagsupermix全式金;goldviewi型核酸染色剂、dtt(二硫苏糖醇)、葡聚糖凝胶、tris、edtana2,,北京索来宝生物科技有限公司;其他试剂均为分析纯。bsa124s电子天平,赛多利斯科学仪器有限公司;chb-100a恒温金属浴,杭州博日科技有限公司;xw-80a旋涡混合器,上海精科实业有限公司;lx-200迷你离心机,海门市其林贝尔仪器;t960型pcr热循环仪,杭州晶格科学仪器有限公司;dyy-6b型稳压稳流电泳仪,北京市六一仪器厂;zf-258全自动凝胶成像仪,上海嘉鹏科技有限公司;nanodrop2000dna浓度测试仪。2.取材方法:取样为:样品取角质层,个子样品由电锯切割横截面,完成由货物样品到片状样品取材,块状样品用刀片刮去外层,以防止外原微生物污染,然后刮取内层样品适量削成细薄片。丝状样品随机取适量,75%乙醇清洗3次,双蒸水冲洗至无醇味,紫外光下照射,晾干。消毒剪剪成碎片,称取适量。3.羊角类和牛角类样品角质层dna的提取方法研究由于角蛋白含有较多的胱氨酸,故二硫键含量特别多,在蛋白质肽链中起交联作用,因此角蛋白化学性质特别稳定,有较高的机械强度。它们不易溶解和消化,常用的动物细胞裂解液无法裂解,预实验时,取适量角质层样品,在加入proteinasek及动物组织裂解缓冲液ga后56℃过夜酶解,样品几乎无变化。ddt(二硫苏糖醇)是一种很强的还原剂,常用于蛋白质中二硫键的还原,可用于阻止蛋白质中的半胱氨酸之间所形成的蛋白质分子内或分子间二硫键。加入proteinasek及动物组织裂解缓冲液ga后56℃酶解3~4小时后,样品裂解情况有较大改善,是影响dna提取完全的关键因素,样品的取样量也是影响dna质量的关键因素。因此,对取样量和dtt的用量进行了单因素考察。3.1取样量的考察称取羚羊角薄片(样品编号1)25mg、40mg、55mg、70mg,加入dtt100µl,proteinasek20µl及裂解缓冲液ga200µl后,56℃金属浴酶解3小时后,按试剂盒提取步骤提取dna,nanodrop2000测定dna的浓度及纯度,结果见表2。表2:取样量考察由表2知,取样量为25mg时,提取的dna纯度较高,取样量增大时,虽然提取浓度也增大,但a260/280值较低,提示有蛋白质污染。进一步pcr扩增也显示,蛋白污染的样品扩增出的条带较暗,确定取样量为25mg。3.2:dtt加入量的考察称取刀片刮取的羚羊角粉(样品编号1)25mg5份,分别加入20µl、40µl、60µl、80µl、100µl的dtt,加入40µl的proteinasek及裂解缓冲液ga400µl后,56℃金属浴酶解,考察样品在10小时内的酶解情况。其结果见表3。表3dtt加入量考察为减少dtt用量,避免浪费,确定dtt加入量为20µl,可使样品在4小时内完全裂解。将上述优选的dna提取方法,取样量25mg,dtt用量20µl,用于所收集的编号为1~10的样品dna提取。提取的dna浓度及纯度结果见表4。表4:编号为1~10的样品dna提取结果3.3所有角类样品的pcr扩增上述所有提取的dna样本(样品编号1~10),用于pcr扩增,扩增引物为co1基因的通用引物,正向引物lco14905′-ggtcaacaaatcataaagatattgg-3′,反向引物hco21985′-taaacttcagggtgaccaaaaaatca-3′,25µl反应体系,模板用量为150~180ng,达不到150~180ng,模板取样定为10-14ul;正向、反向引物各1µl(浓度为10mmol/l);2×easytagsupermix12.5µl;双蒸水补足至25µl。pcr扩增程序为:94℃1min;94℃,1min,45℃,1.5min,72℃,1.5min,5个循环;94℃,1min,50℃,1.5min,72℃,1min,35个循环;72℃,5min。用含有goldviewi型核酸染色剂的1.5%琼脂糖凝胶点样,dl2,000dnamarker为对照,样品5µl与2µl6×loadingbuffer混合均匀点样,tbe缓冲液中电泳,凝胶成像仪成像。割取750bp处的dna条带,胶回收纯化pcr产物,上海桑尼生物公司测序。上述提取样品的pcr结果见图2(点样顺序对应样品编号)。3.4低浓度样品的二次扩增针对图2中点样序号为6、7的样品(对应样品编号为6,7)存在浓度低,无法完成测序,为解决这一问题,重新提取dna,在最终溶解保存dna时,减少溶解dna的te用量,提升浓度。提取的dna第一次扩增后,紫外灯下割取750bp处的条带,切成碎块,100µl双蒸水溶解,60℃温育,取适量,进行二次扩增。模板用量分别为2µl、5µl、10µl;正向、反向引物各1µl(浓度为10mmol/l);2×easytagsupermix12.5µl;双蒸水补足至25µl。扩增程序为95℃1min,40℃,1min,72℃,1.5min,72℃,1.5min,35个循环模,72℃,保存7min。1.5%琼脂糖点样,tbe缓冲液电泳,凝胶成像仪成像,结果见图3。由图3知,二次扩增时模板取样量5µl时,效果最好,可用于测序;10µl模板抑制了pcr反应;2µl模板条带亮度适中。割取750bp处的dna条带,胶回收纯化pcr产物,上海桑尼生物公司测序。测序结果见图4和图5。序列经过与ncbi数据库比对,相似度均为99%以上,确定了具体物种名称,见表5。图1中标注出经过与ncbi数据库比对后确定各样品的物种名称。采用本发明发法,在确定取样量的基础上,加入适当量的dtt,短时间内(4小时内),可使角质层样品完全裂解,保证了dna最大程度提出,也能满足陈旧样品的dna提取,无需加大取样量,无需进口痕量dna提取试剂盒,成功测出所有样品的co1序列,将序列与ncbi数据库上的序列比对,可得出具体物种信息,从比对结果可知,收集的样品除黄牛角比对结果为牦牛角外(可能依据性状鉴定会出现差错),其余样品与比对结果一致,说明收集样品的物种信息是正确的,确定了传统方法不能鉴定的羚羊角丝与羚羊角块。因此本发明建立的dna提取方法,提取dna完全,使提取的dna质量满足常规pcr要求,可标准化操作,可应用于多种动物角的提取。可满足新鲜和陈旧的动物角类样品,同时避免取骨塞部位(牛角类和羊角类样品含有骨塞)时,用edta脱钙预处理,因脱钙次数和时间不好掌握,造成dna的提取不完全或dna被破坏。表5样品比对结果表本发明所述方法对动物角类药材的角质层部位提取动物角中的dna,对于dna严重降解的样本,通过减少溶解dna的溶剂用量,提高初始浓度,或切胶纯化,进行二次扩增,最终使所有样品的pcr反应结果满足测序要求,成功测出所有样品的线粒体co1基因序列。在取材上,样品由于坚硬,手工用刀片刮粉、钢锉锉粉或电动研磨器磨粉,均存在效率低下的缺点,难以完成取样,后改为电锯切割横截面,完成由样品到片状样品取材,片状样品进一步用刀片刮去外层,以防止外原微生物污染。实验例:采用保存10年以上陈旧羊角样品,分别按常规试剂盒处理方法与本发明方法处理,常规试剂盒处理方法不包括第(3)步骤,本发明方法包括第(3)步骤,试剂盒采用天根生化科技有限公司的血液/细胞/组织基因组dna提取试剂盒(离心柱型),目录号:dp304(包含proteinasek,缓冲液ga,缓冲液gb,缓冲液gd,漂洗液pw,吸附柱cb3)。样品信息见表6。表6:存放10年以上的样品信息编号样品名称样品存放时间产地1羚羊角(个子)15年西伯利亚2羚羊角块13年俄罗斯3藏羚羊角(个子)12年新疆具体步骤为:(1)样品前处理:取表6的角类药材样品,先将样品用电锯切割横截面,完成由个子样品到片状样品取材;然后用刀片刮去外层,削取薄片适量,样品编号为1、3,样品编号2的处理:用刀片刮去外层,削取薄片适量;(2)取样:称取刮取的羚羊角薄片30mg于1.5mlep管中,(3)加入dtt30µl,加入proteinasek20µl及裂解缓冲液ga200µl后,56℃金属浴酶解3-4小时,(4)按试剂盒提取步骤提取dna:加入200µl缓冲液gb,充分颠倒混匀,70℃放置10分钟,离心除管盖内壁水珠;加入200µl无水乙醇,充分震荡混匀15秒,离心除去管盖内壁水珠;将所得溶液加入吸附柱cb3中,12000rpm离心30秒,倒掉废液,将吸附柱cb3放回收集管中,向吸附柱cb3加入500µl缓冲液gd,12000rpm离心30秒,倒掉废液,将吸附柱cb3放回收集管中,向吸附柱cb3加入600µl漂洗液pw,12000rpm离心30秒,倒掉废液,将吸附柱cb3放回收集管中;重复加入200µl无水乙醇,充分震荡混匀15秒,离心除去管盖内壁水珠;将吸附柱cb3放回收集管中,12000rpm离心2分钟,倒掉废液。将吸附柱cb3置室温放置数分钟,以晾干吸附材料中残余的漂洗液,将吸附柱cb3转入一个干净的离心管中,向吸附膜的中间部位悬空滴加80µl洗脱缓冲液te,室温放置2-5分钟,12000rpm离心2分钟,将溶液收集到离心管中。(5)dna浓度测定:nanodrop2000测定dna的浓度及纯度,结果见表7,由表7得知,常规方法对放置10年以上的样品,提出的dna模板浓度低,本发明所述方法可明显提高dna浓度。表7:常规方法与本发明方法提取dna的浓度结果常规方法本发明方法样品名称存放时间dna浓度(ng/µl)dna浓度(ng/µl)羚羊角(个子)15年3.518.3羚羊角块13年2.720.2藏羚羊角(个子)12年5.423.5(6)pcr扩增:提取的dna样本(常规方法与本发明方法)进行pcr扩增:扩增引物为co1基因的通用引物,正向引物lco14905′-ggtcaacaaatcataaagatattgg-3′,反向引物hco21985′-taaacttcagggtgaccaaaaaatca-3′,25µl反应体系,模板量为10µl;浓度为10mmol/l的正向、反向引物各1µl;2×easytagsupermix12.5µl;双蒸水补足至25µl;pcr扩增程序为:94℃1min;94℃,1min,45℃,1.5min,72℃,1.5min,5个循环;94℃,1min,50℃,1.5min,72℃,1min,35个循环;72℃,5min;用含有goldviewi型核酸染色剂的1.5%琼脂糖凝胶点样,dl2,000dnamarker为对照,样品5µl与2µl6×loadingbuffer混合均匀点样,tbe缓冲液中电泳,凝胶成像仪成像;结果见图5,由图5知,在取同样量的模板dna时,常规试剂盒方法在750bp处的扩增条带为阴性,而本发明方法扩增出的dna条带明显。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1