一种非诺贝酸的制备方法与流程
本发明属于药物制备领域,具体而言,本发明涉及一种非诺贝酸的制备方法。
背景技术:
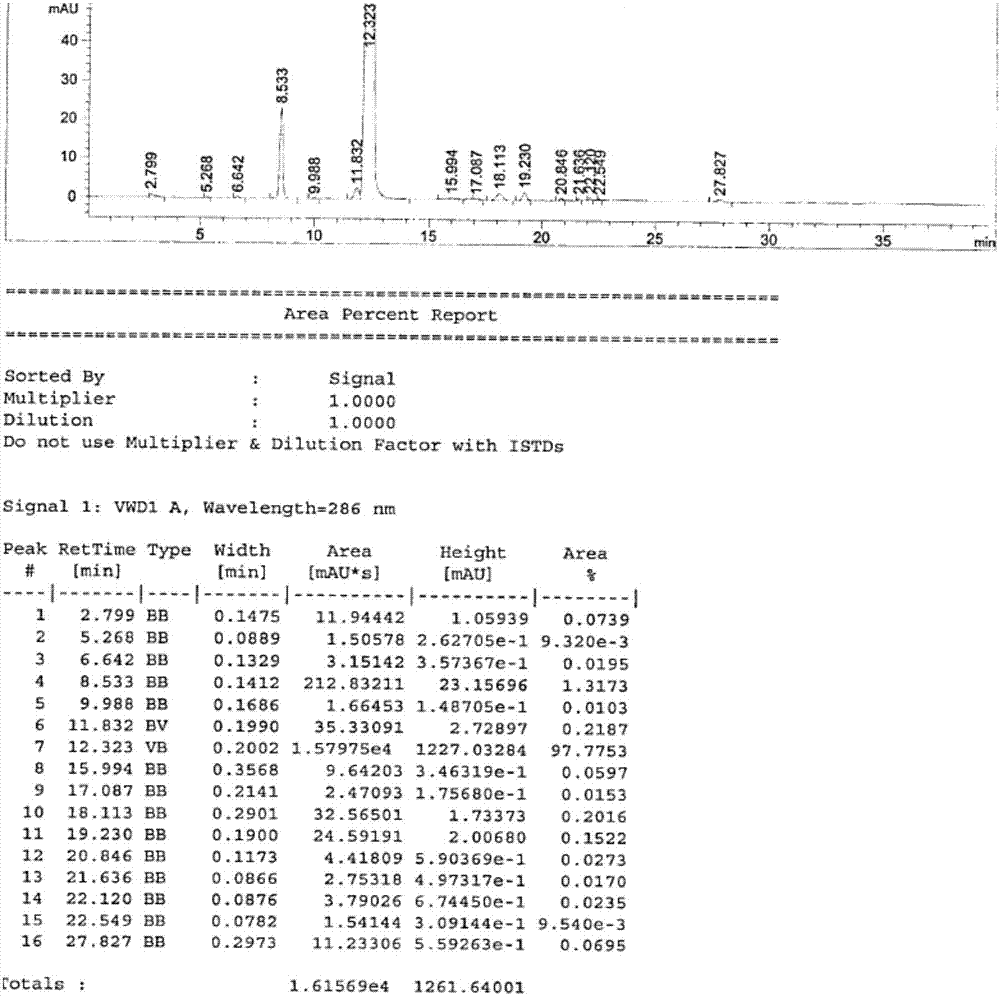
:非诺贝特是目前临床使用较多的苯氧酸酯类降血脂药物之一,相比于非诺贝特,非诺贝酸为其体内代谢的活性物质,其在小肠区内有着极高的溶解度,因此增加了生物利用度,并且其生物利用度不受食物的影响。已公开的资料中对于非诺贝酸制备工艺的报道较少,基本上都是以4-羟基-4’-氯二苯甲酮作为起始物料缩合或者取代产生非诺贝酸。乔德阳,李敢(合成化学,2009)在teba催化合成非诺贝酸中描述了一种以50%氢氧化钾溶液为溶剂,4-羟基-4’-氯二苯甲酮、氯仿、丙酮在相转移催化剂teba的催化下发生缩合反应制备非诺贝酸。中国专利cn103360240a中公布了一种在上述乔德阳等人公布的条件上改进的方法,以四正丁基溴化铵为相转移催化剂,4-羟基-4’-氯二苯甲酮、氯仿、丙酮在碱性条件下发生缩合反应制备非诺贝酸。综上两条路线,虽然均能获得比较满意的收率,但是也存在不少缺陷。缩合反应制备非诺贝酸存在如下缺点:1、使用到昂贵的相转移催化剂teba或者四正丁基溴化铵;2、反应时间长达25h;反应在相转移催化条件下进行,为多相反应,不利于大生产。romand.davis等(synthesis,2004)在improvedmethodforthesynthesisof2-methyl-2-aryloxypropaniocacidderivatives中描述了一种以丁酮为溶剂,4-羟基-4’-氯二苯甲酮、2-溴代异丁酸在氢氧化钠条件下发生取代反应制备非诺贝酸的方法。但是,该取代反应制备非诺贝酸存在如下缺点:1、反应时间长达16~24h;2、反应过程中会有一段时间体系极其粘稠,容易导致搅拌桨电机烧毁,使得大生产存在较大的难度。非诺贝酸具有良好的市场前景,同时目前的工艺报道又都不适合于工业化生产,因此,找到一条适合工业化生产非诺贝酸的工艺就显得非常迫切与必要。技术实现要素:本发明人凭借长期的研究经验与实验探索,找到了一条能够解决上述问题的工艺,既不使用昂贵的相转移催化剂,同时又大大降低了体系的粘稠度,缩短了反应时间,且收率与文献报道相当,更令人意外的是,经此方法制备出的非诺贝酸粗品经精制能够得到纯度不低于99.5%的高纯度非诺贝酸。本发明的目的是提供一种适于产业化的非诺贝酸的制备方法。在本发明的一种实施方案中,本发明提供了一种新的非诺贝酸的制备方法,包括如下步骤:(1)4-氯-4’-羟基二苯甲酮与强碱在丁酮中搅拌混合,加热下,搅拌至少20分钟;(2)向步骤(1)所得到的混合物中加入质子溶剂,搅拌并保温,然后滴加2-溴代异丁酸的丁酮溶液,加毕,反应2-6小时;(3)后处理,经分离得到非诺贝酸粗品;(4)精制,得到非诺贝酸精品。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(1)所述强碱为氢氧化钠、氢氧化钾、叔丁醇钾,优选氢氧化钠。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(1)中所述4-氯-4’-羟基二苯甲酮与强碱的摩尔比为1:2.5~1:8,优选为1:4.5;在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(1)的4-氯-4’-羟基二苯甲酮与步骤(2)的2-溴代异丁酸的摩尔比为1:1~1:2,优选摩尔比为1:1.4。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(1)中所述4-氯-4,-羟基二苯甲酮与丁酮的重量比为1:10~1:20,优选1:15。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(1)所述加热的温度为40~70℃,优选45~60℃。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(2)所述质子溶剂为水、甲醇和乙醇的一种或多种混合溶液,优选水。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(1)所述的4-氯-4’-羟基二苯甲酮与步骤(2)所述的质子溶剂的重量比为1:0.1~1:0.3,优选1:0.2。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(2)所述保温的温度为40~70℃,优选45~60℃。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备工艺,其中,步骤(2)所述反应时间为3~5小时。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(3)所述后处理为:在步骤(2)结束后向体系中加入纯化水洗涤,收集有机相;然后向该有机相中加酸,过滤,水洗滤饼,收集滤饼在40~60℃下真空干燥得到非诺贝酸粗品。更优选地,在步骤(2)结束后向体系中加入纯化水洗涤,收集有机相;然后向该有机相中加酸至ph达到1~3,优选2~3,过滤,水洗滤饼至ph达到4~6,优选5~6,收集滤饼;在40~60℃下真空干燥得到非诺贝酸粗品。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,步骤(3)中所述加入纯化水的量为步骤(1)中所用的4-氯-4’-羟基二苯甲酮重量的4~8倍,优选6倍。在本发明的优选实施方案中,本发明提供的一种新的非诺贝酸的制备方法,其中,所述步骤(4)是:将步骤(3)获得的非诺贝酸粗品溶解于c2~c4的烷醇中,溶解温度65~75℃;向所得溶液中加入活性炭脱色,过滤并收集滤液;将获得的滤液降温,过滤,40~60℃真空干燥得纯度较高的非诺贝酸。这里,优选地,非诺贝酸粗品与c2~c4烷醇的重量比为1:8~1:16,更优选1:9~1:12;所述c2~c4的烷醇为乙醇、正丙醇、异丙醇、正丁醇或异丁醇中的任意一种。进一步优选地,步骤(4)中加入活性炭的重量为步骤(3)中所得的非诺贝酸粗品重量的0.01~0.1倍,更优选0.05倍。进一步优选,将获得的滤液降温是指降温至-10~10℃,更优选5~10℃。与现有技术相比,本发明上述方法不使用相转移催化剂,体系粘稠度小,易于搅拌,反应时间短;制得的非诺贝酸具有99.5%以上的色谱纯度,样品稳定性好,符合要用要求。因此,本发明提供了一种新的非诺贝酸制备工艺,即便于工业生产操作的,产品纯度高、稳定性好的非诺贝酸的制备工艺。附图说明图1表示的是本发明实施例2中的非诺贝酸粗品的hplc图。图2表示的是本发明实施例3a中的非诺贝酸精品的hplc图。图3表示的是本发明实施例3b中的非诺贝酸精品的hplc图。图4表示的是本发明实施例3c中的非诺贝酸精品的hplc图。图5表示的是本发明实施例3d中的非诺贝酸精品的hplc图。图6表示的是本发明实施例3e中的非诺贝酸精品的hplc图。具体实施方式下面通过实施例来具体说明本发明。应该正确理解的是:本发明实施例中的方法仅仅是用于说明本发明而给出的,而不是对本发明的限制,所以,在本发明的方法前体下,对本发明的简单改进均属于本发明的要求保护范围。在本发明中,hplc的分析仪器及方法为:液相色谱仪:agilent1260色谱柱:agilentzorbaxsb-aqc185um4.6*250mm流动相:流动相a为水(用磷酸调节ph值至2.5),流动相b为乙腈。检测波长:286nm。流速:1.0ml/min。柱温:30℃。进样量:10ul洗脱程序:梯度洗脱时间(%)流动相a(%)流动相b(%)05050155050202080302080315050405050实施例1a(非诺贝酸的合成)称取4-氯-4’-羟基二苯甲酮1kg,加入50l干燥的反应釜中,加入12kg丁酮与0.77kg氢氧化钠,搅拌下升温至45~60℃,搅拌30分钟,加入0.2kg纯化水,搅拌1小时,保持45~60℃,向釜中滴加2-溴代异丁酸的丁酮溶液(1kg2-溴代异丁酸溶于3kg丁酮),加毕,反应3~5小时。实施例1b(非诺贝酸的合成)称取4-氯-4’-羟基二苯甲酮1kg,加入50l干燥的反应釜中,加入12kg丁酮与0.77kg氢氧化钠,搅拌下升温至45~60℃,搅拌30分钟,加入0.2kg甲醇,搅拌1小时,保持45~60℃,向釜中滴加2-溴代异丁酸的丁酮溶液(1kg2-溴代异丁酸溶于3kg丁酮),加毕,反应5~6小时。实施例1c(非诺贝酸的合成)称取4-氯-4’-羟基二苯甲酮1kg,加入50l干燥的反应釜中,加入12kg丁酮与0.77kg氢氧化钠,搅拌下升温至45~60℃,搅拌30分钟,加入0.2kg乙醇,搅拌1小时,保持45~60℃,向釜中滴加2-溴代异丁酸的丁酮溶液(1kg2-溴代异丁酸溶于3kg丁酮),加毕,反应5~6小时。实施例2(后处理,分离得到非诺贝酸粗品)在实施例1a的反应结束后,向其中加入6kg纯化水,搅拌,洗涤,分液收集有机相,用1mol/l盐酸调节ph至2~3(室温),析出大量固体,过滤,滤饼用纯化水洗涤至ph至5~6(室温),过滤,收集滤饼,在40~60℃下真空干燥,称量并检测纯度,共得到1.16kg非诺贝酸粗品,hplc检测纯度97%(见附图1)。实施例3a(非诺贝酸粗品的精制)将实施例2获得的非诺贝酸粗品加入20l干燥的反应釜中,加入12kg乙醇,升温至70℃搅拌溶解,溶解条件下加入0.06kg活性炭,搅拌30分钟,过滤,滤液降温至5~10℃,搅拌析晶12小时,过滤,收集滤饼在40~60℃真空干燥,称量并检测纯度,共得到0.98kg非诺贝酸精品,hplc检测纯度99.89%(见附图2)。实施例3b(非诺贝酸粗品的精制)将实施例2获得的非诺贝酸粗品加入20l干燥的反应釜中,加入12kg正丙醇,升温至70℃搅拌溶解,溶解条件下加入0.06kg活性炭,搅拌30分钟,过滤,滤液降温至5~10℃,搅拌析晶12小时,过滤,收集滤饼在40~60℃真空干燥,称量并检测纯度,共得到1.01kg非诺贝酸精品,hplc检测纯度99.91%(见附图3)。实施例3c(非诺贝酸粗品的精制)将实施例2获得的非诺贝酸粗品加入20l干燥的反应釜中,加入12kg异丙醇,升温至70℃搅拌溶解,溶解条件下加入0.06kg活性炭,搅拌30分钟,过滤,滤液降温至5~10℃,搅拌析晶12小时,过滤,收集滤饼在40~60℃真空干燥,称量并检测纯度,共得到1.04kg非诺贝酸精品,hplc检测纯度99.90%(见附图4)。实施例3d(非诺贝酸粗品的精制)将实施例2获得的非诺贝酸粗品加入20l干燥的反应釜中,加入12kg正丁醇,升温至70℃搅拌溶解,溶解条件下加入0.06kg活性炭,搅拌30分钟,过滤,滤液降温至5~10℃,搅拌析晶12小时,过滤,收集滤饼在40~60℃真空干燥,称量并检测纯度,共得到1.02kg非诺贝酸精品,hplc检测纯度99.97%(见附图5)。实施例3e(非诺贝酸粗品的精制)将实施例2获得的非诺贝酸粗品加入20l干燥的反应釜中,加入12kg异丁醇,升温至70℃搅拌溶解,溶解条件下加入0.06kg活性炭,搅拌30分钟,过滤,滤液降温至5~10℃,搅拌析晶12小时,过滤,收集滤饼在40~60℃真空干燥,称量并检测纯度,共得到1.02kg非诺贝酸精品,hplc检测纯度99.78%(见附图6)。对比例称取4-氯-4’-羟基二苯甲酮1kg,加入50l干燥的反应釜中,加入12kg丁酮与0.77kg氢氧化钠,搅拌下升温至45~60℃,搅拌30分钟保持45~60℃,向釜中滴加2-溴代异丁酸的丁酮溶液(1kg2-溴代异丁酸溶于3kg丁酮),加毕,保持45~60℃搅拌反应,随着反应时间的延长,体系粘度越来越大,当反应反应时间在12小时时,由于体系粘度过大,此时电机被烧毁,取样检测,4-氯-4’-羟基二苯甲酮剩余约20%。无法满足要求。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1