一种可进行多物种分子标记的单引物及其标记方法与流程
本发明涉及生物技术领域,特别涉及一种可进行多物种分子标记的单引物及其标记方法。
背景技术:
DNA分子标记是以个体间核苷酸序列变异为基础的遗传标记,直接在DNA水平上检测生物个体之间的差异,能够从DNA水平直接反映生物的遗传本质,是生物个体DNA水平上遗传变异的直接反映。它具有数量丰富、信息量大、多态性高、检测不受季节、环境和个体发育阶段的影响、检测手段简单迅速等优点,已被广泛应用于遗传多样性分析、亲缘关系评价、分子指纹图谱构建、遗传连锁图谱构建、QTL或基因定位与克隆和分子标记辅助选择育种等领域。
自1980年首次提出分子标记的概念以来,有关DNA分子标记的研究不断有文献报道,特别是上世纪90年代以来,发展十分迅速。常规的DNA分子标记技术是使用一对引物进行PCR扩增的,而有一大类DNA分子标记技术只使用一条单引物进行PCR扩增,也被称为基于单引物扩增反应的DNA分子标记技术。单引物扩增反应是指在一个PCR反应中只使用一条单引物,这一条单引物同时充当上游引物和下游引物的作用,当这一条单引物在基因组DNA上的两结合位点之间的距离不是很远的时候,两结合位点之间的序列便可得到有效扩增。自上世纪90年代以来,越来越多的基于单引物扩增反应的DNA分子标记技术被开发出来并得到越来越多地关注和应用,比如RAPD(Williams et al.,1990)、ISSR(Zietkiewicz et al.,1994)、ISTR(Rohde 1996)、IRAP(Kalendaret al.,1999)、IMP(Chang et al.,2001)、SCoT(Collard and Mackill 2009a;熊发前等,2009)、CDDP(Collard and Mackill 2009b;熊发前等,2010b)、iPBS(Kalendar et al.,2010)、HFO-TAG(Levi and Wechter 2010)和CBDP(Singh et al.,2013)。这些基于单引物扩增反应的DNA分子标记技术的技术核心是单引物的设计。其中,RAPD技术中所使用的单引物是完全随机设计的;ISSR技术中所使用的单引物是根据简单重复序列(SSR)来设计的;ISTR技术和IRAP技术中所使用的单引物是根据LTR反转录转座子中的LTR序列来设计的;IMP技术中所使用的单引物是根据MITE转座子中的TIR序列来设计的;SCoT技术中所使用的单引物是根据植物基因中的ATG翻译起始位点侧翼序列的保守性序列来设计的;CDDP技术中所使用的单引物是根据不同物种中同时存在的保守DNA序列或蛋白质序列(如转录因子WRKY、MYB、ERF、KNOX和开花控制MADS-BOX及生长素结合蛋白ABP1)来设计的;iPBS技术中所使用的单引物是根据LTR反转录转座子中毗邻5’LTR序列的保守性的tRNA结合位点(Primer Binding Site,PBS)序列来设计的;HFO-TAG技术中所使用的单引物是根据存在于大量西瓜的Unigenes中的长度为8-10bp的高频率短序列来设计的;CBDP技术中所使用的单引物是根据植物启动子中存在保守一致序列“GGCCAATCT”来设计的。总之,自上世纪九十年代以来,DNA分子标记技术的开发及应用进展十分迅速,新的DNA分子标记技术不断出现,基于单引物扩增反应的DNA分子标记技术也在同步涌现。一般来说,后面出现的DNA分子标记技术大都吸收了前面的优点且克服了它们的部分不足,使之不断优化,但现有的DNA分子标记技术的缺陷仍然存在,如引物设计较复杂、引物数量较少、引物通用性不强、引物退火温度较低、引物扩增重复性较差和引物趋向于非功能编码区域扩增等缺点。
本发明的第一发明人在2015年申请了一项中国专利,专利号为:CN 104946742A,公开了一种针对真核生物基因组中基因外显子区域设计合成的单引物,该引物具有设计简单、合成费用较低、通用性强、利用率高、退火温度较高、扩增重复性较好、产生的多态性较丰富等特点,但该申请并没有公开针对真核生物基因组中基因内含子区域或调控区域设计合成的单引物,但并没有公开利用基于基因内含子区域或调控区域设计的单引物进行多物种分子标记分析的技术。
技术实现要素:
鉴于上述内容,有必要提供一种可进行多物种分子标记的单引物及其标记方法,该引物针对真核生物基因组中基因内含子区域或调控区域设计合成,具有设计简单、合成费用较低、通用性强、利用率高、扩增效率高和产生的多态性丰富等特点。
为达到上述目的,本发明所采用的技术方案是:
一种可进行多物种分子标记的单引物,所述单引物根据真核生物基因组中基因内含子区域或调控区域富含AT设计,单引物序列长度为17bp;所述引物序列由核心序列和选择性碱基序列组成,所述核心序列由从5’端到3’端的14个碱基组成,包括填充序列和核心区序列,其中,从5’端到3’端的第1-第6个碱基为填充序列,从5’端到3’端的第7-第14个碱基为核心区序列;所述选择性碱基由3个碱基组成,为从5’端到3’端的第15-第17个碱基;所述填充序列为富含GC碱基的任意序列;所述核心区序列由AT碱基随机排列组成;所述选择性碱基序列为任意碱基序列;
所述单引物序列为Mem1-Mem32中的任意一条:
Mem1:5’-GCCGGCATTATTATCCA-3’;
Mem2:5’-GCCGGCATTATTATCCC-3’;
Mem3:5’-GCCGGCATTATTATCCG-3’;
Mem4:5’-GCCGGCATTATTATCCT-3’;
Mem5:5’-GCCGGCATTATTATCGA-3’;
Mem6:5’-GCCGGCATTATTATCGC-3’;
Mem7:5’-GCCGGCATTATTATCGG-3’;
Mem8:5’-GCCGGCATTATTATCGT-3’;
Mem9:5’-GCCGGCATTATTATGGA-3’;
Mem10:5’-GCCGGCATTATTATGGC-3’;
Mem11:5’-GCCGGCATTATTATGGG-3’;
Mem12:5’-GCCGGCATTATTATGGT-3’;
Mem13:5’-GCCGGCATTATTATGCA-3’;
Mem14:5’-GCCGGCATTATTATGCC-3’;
Mem15:5’-GCCGGCATTATTATGCG-3’;
Mem16:5’-GCCGGCATTATTATGCT-3’;
Mem17:5’-GGCGCCAATAATAACCA-3’;
Mem18:5’-GGCGCCAATAATAACCC-3’;
Mem19:5’-GGCGCCAATAATAACCG-3’;
Mem20:5’-GGCGCCAATAATAACCT-3’;
Mem21:5’-GGCGCCAATAATAACGA-3’;
Mem22:5’-GGCGCCATTATTATCGC-3’;
Mem23:5’-GGCGCCAATAATAACGG-3’;
Mem24:5’-GGCGCCAATAATAACGT-3’;
Mem25:5’-GGCGCCAATAATAAGGA-3’;
Mem26:5’-GGCGCCAATAATAAGGC-3’;
Mem27:5’-GGCGCCAATAATAAGGG-3’;
Mem28:5’-GGCGCCAATAATAAGGT-3’;
Mem29:5’-GGCGCCAATAATAAGCA-3’;
Mem30:5’-GGCGCCAATAATAAGCC-3’;
Mem31:5’-GGCGCCAATAATAAGCG-3’;
Mem32:5’-GGCGCCAATAATAAGCT-3’。
进一步的,所述单引物适用于真核生物的分子标记分析。
更进一步的,所述真核生物为花生、水稻、玉米、甘蔗、木薯、菜心和真菌中的任意一种。
进一步的,所述引物的解链温度为51℃-61℃。
本发明还提供了一种可进行多物种分子标记的标记方法,该方法包括如下步骤:
(1)按所述序列合成单引物;
(2)提取真核生物的基因组DNA,得到基因组DNA模板;
(3)利用步骤(1)的单引物与步骤(2)的基因组DNA模板在PCR扩增反应体系中进行PCR扩增,得到PCR扩增产物;
(4)将步骤(3)的PCR扩增产物通过琼脂糖凝胶电泳进行分离,再通过溴化乙锭染色,在凝胶成像系统下拍照、保存图像和记录结果。
进一步的,所述PCR扩增反应体系为:浓度为50ng/ul的DNA模板1μl;10×PCR Buffer2μl;浓度为2.5mmol/L的dNTPs 0.5μl;浓度为10pmol/μl的单引物1μl;酶活力单位为5U/μl的Taq酶0.2μl;ddH2O15μl。
进一步的,所述PCR扩增程序为:94℃预变性4min;94℃变性30s,35℃退火1min,72℃延伸90s,总共35个循环;最后72℃延伸10min,4℃保存。
进一步的,所述琼脂糖凝胶的质量浓度为1.5%。
本发明具有如下有益效果:
1、本发明中所提供的单引物是根据真核生物基因组中基因内含子区域或者调控区域富含AT来设计的,设计简单,可以在真核生物间通用,通用性强,大大降低了合成费用,也大大地提高了利用率;另外,引物扩增效率高和产生的多态性丰富;除此之外,本发明中的所提供的单引物只要根据所述原理来设计,就可以大批量设计出,从而方便各种研究。
2、本发明的分子标记方法是建立在PCR反应的基础上的,该方法简单高效、扩增效率高、扩增条带丰富、产生的多态性高、结果可靠、条带易于分离及可测序,不需使用同位素。
3、本发明中所提供的一种可进行多物种分子标记的分子标记方法,虽然像RAPD、ISSR一样,都是基于单引物扩增反应的,但该方法中所使用的单引物是根据真核生物基因组中基因内含子区域或者调控区域富含AT来设计合成的,PCR扩增时一条单引物可同时锚定结合在基因组中的基因内含子区域中的两个位点,产生的扩增产物包括内含子区域甚至临近的外显子区域,产生的多态性标记多来始于内含子长度多态性,与基因紧密相关,为功能型标记,方便进行分子标记辅助选择育种。
【附图说明】
图1是本发明的一种可进行多物种分子标记的标记方法示意图。
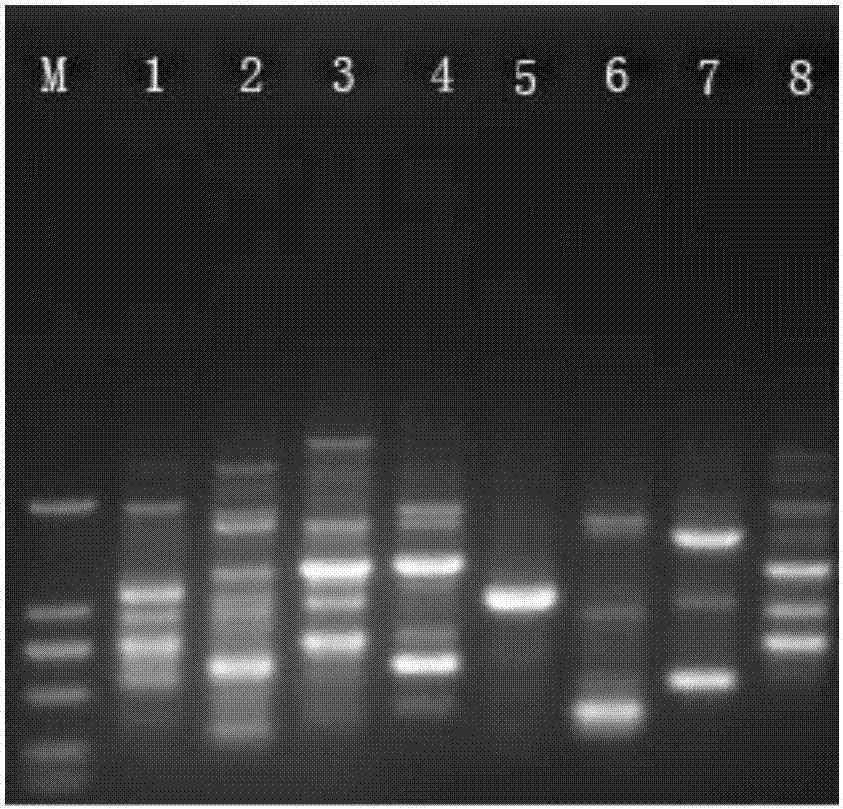
图2是以花生的基因组DNA为模板对其中的8条单引物的扩增有效性进行验证和筛选的电泳图;
其中,M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-8分别为Mem1、Mem2、Mem3、Mem4、Mem5、Mem6、Mem7和Mem8。
图3是以甘蔗的基因组DNA为模板对其中的8条单引物的扩增有效性进行验证和筛选的电泳图;
其中,M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-8分别为Mem1、Mem2、Mem3、Mem4、Mem5、Mem6、Mem7和Mem8。
图4是单引物Mem2在16种水稻材料中的扩增电泳图;
其中M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-16分别为Kasalah、白皮糯谷、晚稻糯、花谷、太晚白、细谷、黄白占、广恢998、日本晴、黄红糯、香糯、山粳、马来、花皮、晚旱稻和荣怕红的扩增电泳图。
图5是单引物Mem30在11种玉米材料中的扩增电泳图;
其中M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-11分别为正大619、兆丰788、桂单688、桂单591、兆丰688、桂单0810、桂单589、云瑞88、玉美头105、南校9665和桂单901。
图6是单引物Mem22在24种甘蔗材料中的扩增电泳图;
其中,M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-24分别为桂糖11、桂糖21、新台糖22、桂糖29、桂糖31、桂糖32、桂糖36、桂糖37、桂糖40、桂糖41、桂糖42、桂糖43、桂糖44、桂糖45、桂糖46、桂糖47、桂糖48、桂糖49、桂糖98-296、桂糖03-2287、桂糖07-108、桂糖08-460、桂糖08-1180和拔地拉。
图7是单引物Mem5在5种木薯材料中的扩增电泳图;
其中M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-5分别为华南205、华南9号、南植199、新选048和col22的扩增电泳图。
图8是单引物Mem2在13种菜心材料中的扩增电泳图;
其中,M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-13分别为特青60天粗条菜心、澳洲008全年油绿甜菜心、澳洲超级608、香港45天油青甜菜心、桂柳十月柳叶菜心、绿宝701、3-1菜心、3-6-1菜心、5-3菜心、5-4菜心、5-14菜心、6-6菜心和6-6-1菜心。
图9是单引物Mem2在4种真菌材料中的扩增电泳图;
其中,M为DL2000Marker,条带从上到下依次为2000bp、1000bp、750bp、500bp、250bp和100bp;编号1-4分别为镰刀菌、毛霉、曲霉和白僵菌。
【具体实施方式】
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。
引物设计:
本发明可进行多物种分子标记的单引物,根据真核生物基因组中基因内含子区域或调控区域富含AT设计,单引物序列长度为17bp;所述引物序列由核心序列和选择性碱基序列组成,所述核心序列由从5’端到3’端的14个碱基组成,包括填充序列和核心区序列,其中,从5’端到3’端的第1-第6个碱基为填充序列,从5’端到3’端的第7-第14个碱基为核心区序列;所述选择性碱基由3个碱基组成,为从5’端到3’端的第15-第17个碱基;所述填充序列为富含GC碱基的任意序列;所述核心区序列由AT碱基随机排列组成;所述选择性碱基序列为任意碱基序列。引物序列、引物AT含量和解链温度Tm如下表1所示:
表1实施例中的引物序列信息表
实施例1:本实施例以花生为样品进行单引物扩增有效性验证和筛选实验
1、样品材料:花生新品系桂花911(暂名)。
2、花生基因组DNA提取:取新鲜花生嫩叶样品0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0mlCTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用600μl DEPC水充分溶解沉淀,再加入600μl Tris平衡酚充分混匀抽提,静置5min,冷冻离心机中以转速为12000rpm离心10min,取上清液并在上清液中加入与上清液等体积的事先预冷的无水乙醇于-40℃冰箱中沉淀30min,再次用白色小枪头或牙签将白色沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,短时间(5min以内)晾干后,用200μl DEPC水或TE溶解沉淀。
3、PCR扩增反应:以花生的DNA为模版,对单引物进行验证和筛选:
①PCR扩增反应体系为:50ng的DNA模板1μl;10×PCR Buffer 2μl;浓度为2.5mmol/L的dNTPs溶液0.5μl;浓度为10pmol/μl的单引物1μl;酶活力单位为5U/μl的Taq酶0.2μl;蒸馏15μl水。
②PCR扩增反应程序为:94℃预变性4min;94℃变性30s,50℃退火1min,72℃延伸90s,总共35个循环;最后72℃延伸10min,4℃保存。
4、电泳检测:
首先制备浓度为1.5%的琼脂糖凝胶:称取取1.5g琼脂糖置于锥形瓶中,加入100ml的1×TAE缓冲液并在微波炉中进行加热煮沸直至琼脂糖全部融化、摇匀得到浓度为1.5%的琼脂糖凝胶液,待胶凝固后,按照上述的PCR反应体系和反应程序进行PCR扩增,PCR产物中加入上样缓冲液并点样进行电泳,电泳完成后,用溴化乙锭进行染色,在凝胶成像系统下观察、拍照,检测结果如图2所示(引物Mem1-Mem32都能通过上述方法进行验证,本实施例的图仅公开Mem1-Mem8的扩增电泳图)。
实施例2:本实施例以甘蔗为样品进行单引物扩增有效性验证和筛选实验
1、样品材料:甘蔗品种新台糖22。
2、甘蔗基因组DNA提取:取新鲜甘蔗芯叶0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0mlCTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用200μl DEPC水充分溶解沉淀。
3、PCR扩增反应:其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图3所示(引物Mem1-Mem32都能通过上述方法进行验证,本实施例的图仅公开引物Mem1-Mem8的扩增电泳图)。
实施例3:本实施例以水稻为样品进行分子标记分析
1、样品材料:16个水稻品种,分别为:Kasalah、白皮糯谷、晚稻糯、花谷、太晚白、细谷、黄白占、广恢998、日本晴、黄红糯、香糯、山粳、马来、花皮、晚旱稻和荣怕红。
2、基因组DNA提取:取新鲜水稻嫩叶0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0mlCTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用200μl DEPC水充分溶解沉淀。
3、PCR扩增反应:以上述16个水稻样本的DNA为模版,利用单引物Mem2进行扩增(本实施例仅举一个例子进行说明,但本发明的单引物Mem1-Mem32都能实现上述扩增),其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图4所示。
实施例4:本实施例以玉米为样品进行分子标记分析
1、样品材料:11个玉米品种,分别为:正大619、兆丰788、桂单688、桂单591、兆丰688、桂单0810、桂单589、云瑞88、玉美头105、南校9665和桂单901。
2、基因组DNA提取:取新鲜玉米嫩叶样品0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0mlCTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用200μl DEPC水充分溶解沉淀。
3、PCR扩增反应:以上述11个玉米样本的DNA为模版,利用单引物Mem30进行扩增(本实施例仅举一个例子进行说明,但本发明的单引物Mem1-Mem32都能实现上述扩增),其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图5所示。
实施例5:本实施例以甘蔗为样品进行分子标记分析
1、样品材料:24个甘蔗品种,分别为:桂糖11、桂糖21、新台糖22、桂糖29、桂糖31、桂糖32、桂糖36、桂糖37、桂糖40、桂糖41、桂糖42、桂糖43、桂糖44、桂糖45、桂糖46、桂糖47、桂糖48、桂糖49、桂糖98-296、桂糖03-2287、桂糖07-108、桂糖08-460、桂糖08-1180和拔地拉。
2、基因组DNA提取:取新鲜甘蔗嫩叶样品0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0mlCTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用200μl DEPC水充分溶解沉淀。
3、PCR扩增反应:以上述24个甘蔗样本的DNA为模版,利用单引物Mem22进行扩增(本实施例仅举一个例子进行说明,但本发明的单引物Mem1-Mem32都能实现上述扩增),其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图6所示。
实施例6:本实施例以木薯为样品进行分子标记分析
1、样品材料:5个木薯品种,分别为:华南205、华南9号、南植199、新选048和col22。
2、基因组DNA提取:取新鲜木薯嫩叶0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0ml CTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用600μl DEPC水充分溶解沉淀,再加入600μl Tris平衡酚充分混匀抽提,静置5min,冷冻离心机中以转速为12000rpm离心10min,取上清液并在上清液中加入与上清液等体积的事先预冷的无水乙醇于-40℃冰箱中沉淀30min,再次用白色小枪头或牙签将白色沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,短时间(5min以内)晾干后,用200μl DEPC水或TE溶解沉淀。
3、PCR扩增反应:以上述5个木薯样本的DNA为模版,利用单引物Mem5进行扩增(本实施例仅举一个例子进行说明,但本发明的单引物Mem1-Mem32都能实现上述扩增),其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图7所示。
实施例7:本实施例以菜心为样品进行分子标记分析:
1、样品材料:13个菜心品种,分别为:特青60天粗条菜心、澳洲008全年油绿甜菜心、澳洲超级608、香港45天油青甜菜心、桂柳十月柳叶菜心、绿宝701、3-1菜心、3-6-1菜心、5-3菜心、5-4菜心、5-14菜心、6-6菜心、6-6-1菜心。
2、基因组DNA提取:取新鲜菜心嫩叶样品0.3-0.5g,用剪刀剪碎,放入研钵中并在研钵中加入1g的PVP,分多次倒入足量的液氮,将叶片样品充分研磨成粉末状,将全部粉末样品转移到事先加入了1.0mlCTAB提取液且经过65℃预热的2ml离心管中,再加入20μlβ-巯基乙醇,于65℃水浴锅中温浴60min,期间上下颠倒混匀,以便DNA充分析出;然后在冷冻离心机中以转速为12000rpm离心10min后除去底部残渣,吸取上清液到另外一支2ml离心管,并在上清液中加入0.5μl的RNaseA,上下充分颠倒混匀,静置5min,加入与上清液等体积的氯仿抽提,上下充分颠倒混匀,静置5min;然后在冷冻离心机中以转速为12000rpm离心10min,吸取上清液并加入与上清液等体积且事先在-40℃预冷的无水乙醇于-40℃冰箱中沉淀30min,用白色小枪头或牙签将沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,再用600μl DEPC水充分溶解沉淀,再加入600μl Tris平衡酚充分混匀抽提,静置5min,冷冻离心机中以转速为12000rpm离心10min,取上清液并在上清液中加入与上清液等体积的事先预冷的无水乙醇于-40℃冰箱中沉淀30min,再次用白色小枪头或牙签将白色沉淀挑出来到新的1.5ml离心管中,用75%vol乙醇洗涤沉淀一遍,短时间(5min以内)晾干后,用200μl DEPC水或TE溶解沉淀。
3、PCR扩增反应:以上述13个菜心样本的DNA为模版,利用单引物Mem2进行扩增(本实施例仅举一个例子进行说明,但本发明的单引物Mem1-Mem32都能实现上述扩增),其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图8所示。
实施例8:本实施例以真菌为样品进行分子标记分析
1、样品材料:4种真菌,分别为:镰刀菌、毛霉、曲霉和白僵菌。
2、基因组DNA提取::取一粒常温保存的菌核放到加有硫酸链霉素的PDA培养基平板上,28℃培养两天,用口径5mm的打孔器打成菌饼,挑取两块菌饼接种到装有100mL PSB培养基(马铃薯200g、蔗糖20g和水1000mL)的三角瓶中,28℃培养3-5天,在产生菌核前收集菌丝体,用无菌水冲洗2次,无菌条件下风干,再将干燥的新鲜菌丝体放入加有少许经灭菌的石英砂的研钵中,加入液氮后充分研磨,充分研磨后转移到1.5mL离心管中,加入事先在65℃预热的600μL CTAB提取液,振荡混匀后放入水浴锅中,65℃恒温水浴30min,每10min振荡混匀一次,接着将提取液在冷冻离心机中以转速为12000rpm离心10min后取上清液转入另一支1.5ml离心管中,加与上清液等体积的氯仿/tris饱和酚/异戊醇混合液[V(氯仿):V(tris饱和酚):V(异戊醇)=24:24:1]充分摇匀,于冷冻离心机中以转速为12000r/min离心10min,取上清液移入另一离心管中,加入上清液2.5倍体积且经-30℃预冷的无水乙醇和上清液10%体积的3mol/L NaAc溶液(pH 6.0)沉淀,于冷冻离心机中以转速为12000r/min离心10min,弃上清液,将沉淀用70%vol的酒精洗2~3次,晾干,加200μL TE(含10mmol/L Tris-HCl和1mmol/L EDTA pH8.0)缓冲液充分溶解后加入RNaseA于水浴锅中37℃恒温酶解1h。重复用氯仿/Tris饱和酚/异戊醇混合液抽提1-2次,直至中间层无明显沉淀,移取上清液至一新的离心管中,加上清液2.5倍体积且在-20℃预冷的无水乙醇和上清液10%体积的3mol/L NaAc溶液(pH 6.0)沉淀,于冷冻离心机中以转速为12000r/min离心10min,弃上清液,用70%vol的酒精洗沉淀2~3次,晾干,加50μL TE(含10mmol/L Tris-HCl和1mmol/L EDTA pH8.0)缓冲液充分溶解后-20℃保存。
3、PCR扩增反应:以上述4个真菌品种的DNA为模版,利用单引物Mem2进行扩增(本实施例仅举一个例子进行说明,但本发明的单引物Mem1-Mem32都能实现上述扩增),其中,PCR扩增反应体系和反应程序与实施例1相同。
4、电泳检测方法与实施例1相同,检测结果如图9所示。
验证及结果分析:
1、数据采集与分析:根据计算机软件分析的要求,对实施例3-8的琼脂糖凝胶电泳的胶图上的条带进行记录和统计,将每一条带视为一个位点,有条带的记为1(强带和弱带均记为1),无条带的记为0,从而形成0/1数字矩阵,把该数字矩阵输入计算机软件当中,统计扩增总条带数和多态性条带数,计算多态性比例。
2、结果:由图4-图9中显示的扩增电泳结果可见:采用本发明提供的一种可进行多物种分子标记的标记方法,可以从水稻、玉米、甘蔗、木薯、菜心和真菌等多种真核生物中获得丰富的多态性条带,条带清晰工整,揭示物种内不同材料间的遗传差异,结果可靠稳定。由图4、图8和图9中显示的扩增电泳结果还可以看出:同一条单引物(Mem2)在不同物种材料中(水稻、菜心和真菌)扩增产生了不同的条带模式,说明本发明中的单引物具有在不同真核生物间使用的通用性。由图2和图3中显示的扩增电泳结果可以看出,不同单引物(Mem1-Mem8)在同一种材料中也产生了不同的扩增条带模式,这证明了单引物扩增的有效性。因此本发明方法是一种极为稳定可靠有效的基于单引物扩增反应的DNA分子标记方法。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明的保护范围应以所附权利要求为准。
SEQUENCE LISTING
<110> 广西壮族自治区农业科学院经济作物研究所
<120> 一种可进行多物种分子标记的单引物及其标记方法
<130> 2016
<160> 32
<170> PatentIn version 3.5
<210> 1
<211> 17
<212> DNA
<213> 人工序列
<223> Mem1
<400> 1
gccggcatta ttatcca 17
<210> 2
<211> 17
<212> DNA
<213> 人工序列
<223> Mem2
<400> 2
gccggcatta ttatccc 17
<210> 3
<211> 17
<212> DNA
<213> 人工序列
<223> Mem3
<400> 3
gccggcatta ttatccg 17
<210> 4
<211> 17
<212> DNA
<213> 人工序列
<223> Mem4
<400> 4
gccggcatta ttatcct 17
<210> 5
<211> 17
<212> DNA
<213> 人工序列
<223> Mem5
<400> 5
gccggcatta ttatcga 17
<210> 6
<211> 17
<212> DNA
<213> 人工序列
<223> Mem6
<400> 6
gccggcatta ttatcgc 17
<210> 7
<211> 17
<212> DNA
<213> 人工序列
<223> Mem7
<400> 7
gccggcatta ttatcgg 17
<210> 8
<211> 17
<212> DNA
<213> 人工序列
<223> Mem8
<400> 8
gccggcatta ttatcgt 17
<210> 9
<211> 17
<212> DNA
<213> 人工序列
<223> Mem9
<400> 9
gccggcatta ttatgga 17
<210> 10
<211> 17
<212> DNA
<213> 人工序列
<223> Mem10
<400> 10
gccggcatta ttatggc 17
<210> 11
<211> 17
<212> DNA
<213> 人工序列
<223> Mem11
<400> 11
gccggcatta ttatggg 17
<210> 12
<211> 17
<212> DNA
<213> 人工序列
<223> Mem12
<400> 12
gccggcatta ttatggt 17
<210> 13
<211> 17
<212> DNA
<213> 人工序列
<223> Mem13
<400> 13
gccggcatta ttatgca 17
<210> 14
<211> 17
<212> DNA
<213> 人工序列
<223> Mem14
<400> 14
gccggcatta ttatgcc 17
<210> 15
<211> 17
<212> DNA
<213> 人工序列
<223> Mem15
<400> 15
gccggcatta ttatgcg 17
<210> 16
<211> 17
<212> DNA
<213> 人工序列
<223> Mem16
<400> 16
gccggcatta ttatgct 17
<210> 17
<211> 17
<212> DNA
<213> 人工序列
<223> Mem17
<400> 17
ggcgccaata ataacca 17
<210> 18
<211> 17
<212> DNA
<213> 人工序列
<223> Mem18
<400> 18
ggcgccaata ataaccc 17
<210> 19
<211> 17
<212> DNA
<213> 人工序列
<223> Mem19
<400> 19
ggcgccaata ataaccg 17
<210> 20
<211> 17
<212> DNA
<213> 人工序列
<223> Mem20
<400> 20
ggcgccaata ataacct 17
<210> 21
<211> 17
<212> DNA
<213> 人工序列
<223> Mem21
<400> 21
ggcgccaata ataacga 17
<210> 22
<211> 17
<212> DNA
<213> 人工序列
<223> Mem22
<400> 22
ggcgccatta ttatcgc 17
<210> 23
<211> 17
<212> DNA
<213> 人工序列
<223> Mem23
<400> 23
ggcgccaata ataacgg 17
<210> 24
<211> 17
<212> DNA
<213> 人工序列
<223> Mem24
<400> 24
ggcgccaata ataacgt 17
<210> 25
<211> 17
<212> DNA
<213> 人工序列
<223> Mem25
<400> 25
ggcgccaata ataagga 17
<210> 26
<211> 17
<212> DNA
<213> 人工序列
<223> Mem26
<400> 26
ggcgccaata ataaggc 17
<210> 27
<211> 17
<212> DNA
<213> 人工序列
<223> Mem27
<400> 27
ggcgccaata ataaggg 17
<210> 28
<211> 17
<212> DNA
<213> 人工序列
<223> Mem28
<400> 28
ggcgccaata ataaggt 17
<210> 29
<211> 17
<212> DNA
<213> 人工序列
<223> Mem29
<400> 29
ggcgccaata ataagca 17
<210> 30
<211> 17
<212> DNA
<213> 人工序列
<223> Mem30
<400> 30
ggcgccaata ataagcc 17
<210> 31
<211> 17
<212> DNA
<213> 人工序列
<223> Mem31
<400> 31
ggcgccaata ataagcg 17
<210> 32
<211> 17
<212> DNA
<213> 人工序列
<223> Mem32
<400> 32
ggcgccaata ataagct 17
- 还没有人留言评论。精彩留言会获得点赞!
- 一种荧光定量PCR技术检测意大利蜜蜂王浆主蛋白MRJP 2基因表达的方法与流程
- 一种用于对不同银杏进行基因分型的SNP引物及检测方法与流程
- 用于基因甲基化检测的引物和探针、采样方法、试剂盒与流程
- 一种基于荧光PCR检测ESR1基因突变的引物及探针的制造方法与工艺
- 转基因甜菜GTSB77品系特异性实时荧光PCR检测引物、探针、方法和试剂盒与流程
- 一种基于单核苷酸多态性的葡萄球菌种水平鉴定和菌株分型一体化方法与流程
- 一套毛花猕猴桃和中华猕猴桃物种间杂交子代的鉴定引物的制造方法与工艺
- 检测黄杉大小蠹的引物、试剂盒及检测方法与流程
- 环介导等温扩增技术检测铜绿假单胞菌的引物和试剂盒的制造方法与工艺
- 一种用于鉴定浙贝母的引物对及其应用的制造方法与工艺