一种环糊精‑喜树碱类超分子化疗药物及其制备和应用的制作方法
本发明涉及生物医药技术、纳米医药、超分子化学及药物控制释放技术领域,具体地说是一种具有肿瘤特异性还原降解释药的环糊精-喜树碱类前药的制备方法和应用。
背景技术:
癌症又名为恶性肿瘤,已成为威胁人类生存健康的主要原因,是全球第二大死亡原因。据世界卫生组织统计,2015年癌症造成约880万人死亡,占全球死亡率的六分之一。由于诊断和治疗资源的匮乏,其中约70%的癌症死亡发生在中低收入国家。治疗癌症、提高癌症病人的生存率和生活质量成为了一个世界性的难题。目前,化疗手段仍然是癌症治疗的主要方法之一。然而,大多数化疗药物,比如喜树碱(camptothecin)、紫杉醇(paclitaxel)等,在水中溶解度有限,稳定性差,而且对人体正常组织具有严重的副作用。纳米药物由于其独特的性质被科学家们广泛的用于癌症研究和治疗。纳米药物可以有效提高抗癌药物在生理环境中的分散性和稳定性,延长纳米药物在血液中的循环时间,提高药物在肿瘤组织中的富集。相比较于传统的小分子化疗药物,纳米药物可以有效提高抗肿瘤活性,降低对正常组织的毒副作用。因此,设计有效的纳米载药体系一直是药物控制释放领域的研究焦点。
喜树碱(camptothecin,cpt)是一种细胞毒性喹啉类生物碱,能抑制dna拓扑异构酶i(topoi),在临床前研究中具有显著的抗肿瘤活性。但由于其自身水溶性差,生理环境下易水解失活以及严重的毒副作用,喜树碱没能在临床试验中获得成功。通过对喜树碱改性,目前已有几个喜树碱类似物药物通过了美国食品和药物管理局(fda)批准用于结肠癌的治疗,如伊立替康(irinotecan)和拓扑替康(topotecan)。另一方面,这些药物中的内酯结构(lactoneform)在血液循环过程中容易水解成羧基结构,从而导致药效急剧下降。因此非常有必要开发出一种具有肿瘤选择性释放的纳米药物来投递喜树碱。
技术实现要素:
本发明的首要目的是提供一种喜树碱类-环糊精前药,在水溶液中可以通过环糊精与喜树碱之间的输水相互作用力自组装形成纳米粒子,实现降低对正常组织毒性的同时提高抗肿瘤活性。
本发明的另一目的是提供制备所述的喜树碱类-环糊精前药的方法。
本发明的再一目的是提供所述的前药在制备癌症治疗药物中的应用。
本发明的上述目的通过以下技术方案实现:
首先,本发明提供一种喜树碱-环糊精前药,它是喜树碱或其衍生物和环糊精形成的超分子前药,其结构如式(i)所示:
其中,n1取1-3的整数,即所述的环糊精可以选用α-环糊精、β-环糊精或者γ-环糊精的一种;
r1为
r2为
r3为
r4为喜树碱或其衍生物基团;
x为s或者-ch2-中的一种;
n2、n3和n4为重复单元数,均取0-10的整数;优选0-5的整数;进一步优选0-2的整数。
本发明优选的方案中,所述的式(i)中的r4来自式(ii)-式(v)所示结构中的一种;进一步优选来自式(ii)或式(iii)所示的结构;最优选来自式(ii)所示的结构:
式(ii)为喜树碱的化学结构式,式(iii)为7-乙基-10-羟基喜树碱化学结构式,式(iv)为伊立替康的化学结构式,式(v)为拓扑替康的化学结构式。
本发明的方案中,所述的r1优选为
本发明的方案中,所述的r2优选为
本发明的方案中,所述的r3最优选
本发明进一步优选的方案中,所述的喜树碱类-环糊精前药是喜树碱和β-环糊精形成的超分子前药,即所述式(i)中的n1=2;r1为
本发明最优选的所述喜树碱类-环糊精前药,是结构如下式(vi)~(xiii)所示的任意一种:
本发明还提供一种制备喜树碱类-环糊精前药的方法,是以喜树碱类为原料,在有机溶剂中,通过与带有相应基团的原料反应,制成喜树碱类前药中间体,最后所述中间体再和环糊精反应制备得到所述的喜树碱类-环糊精前药。
本发明的一种具体制备方法包括以下步骤:
1)在有机溶剂中,在酰化催化剂存在下使用三光气活化喜树碱类内酯环上的羟基,再加入过量的2,2'-二硫代二乙醇或1,6-己二醇反应,得到中间体醇;
2)在有机溶剂中,对步骤1)得到的中间体醇进行活化,使中间体醇直接转化为活化酯;
或者,
在有机溶剂中,先在催化剂存在下使用丁二酸酐将步骤1)得到的中间体醇转化为羧酸封端的中间体酸,再在活化剂存在下对所述中间体酸进行羰基活化得到活化酯;
3)步骤2)得到的活化酯再和环糊精衍生物在有机溶剂中混合,并加入催化剂搅拌反应,得到所述的喜树碱类-环糊精前药。
本发明所述制备方法中,步骤1)所述的有机溶剂可以选自二氯甲烷、氯仿、四氢呋喃、1,4-二氧六环或二甲基甲酰胺中的任意一种;优选二氯甲烷。步骤1)所述的酰化催化剂可以选自4-(二甲基氨基)吡啶、三乙胺或n,n-二异丙基乙胺中的任意一种;优选4-(二甲基氨基)吡啶。
本发明所述制备方法中,步骤1)所述的喜树碱类优选喜树碱或7-乙基-10-羟基喜树碱;最优选喜树碱。
本发明所述制备方法中,步骤2)所述的中间体醇的活化,优选使用羰基咪唑或炔丁酸与所述的中间体醇在二甲亚砜中反应。
本发明所述制备方法中,步骤2)所述的中间体酸的羰基活化,优选使用n-羟基琥珀酰亚胺与所述的中间体酸在无水二氯甲烷中反应。
本发明所述制备方法中,步骤2)所述的催化剂可以选自4-(二甲基氨基)吡啶、三乙胺或n,n-二异丙基乙胺中的任意一种,优选4-(二甲基氨基)吡啶;步骤2)所述的活化剂优选1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐。
本发明所述制备方法中,步骤3)所述的环糊精衍生物选自α-环糊精、β-环糊精、γ-环糊精或它们的衍生物中的任意一种;优选β-环糊精或其衍生物中的任意一种;进一步优选6-乙氨基-β-环糊精、6-丁氨基-β-环糊精、6-己氨基-β-环糊精或6-叠氮-β-环糊精中的任意一种。
本发明所述制备方法中,步骤3)所述的有机溶剂选自二甲基甲酰胺或二甲基亚砜中的任意一种;优选二甲基甲酰胺;步骤3)所述的催化剂选自三乙胺、4-(二甲基氨基)吡啶或n,n-二异丙基乙胺中的任意一种,优选三乙胺。
本发明所述制备方法优选的一种实施方式中,制备了式(vi)、(xi)和(xii)所示的前药,其合成路线如下:
具体包括如下步骤:在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用三光气活化喜树碱的羟基,然后与过量的2,2'-二硫代二乙醇反应,制备得到中间体醇1;然后在无水二氯甲烷中在4-二甲氨基吡啶存在下使用丁二酸酐将中间体醇1转化为中间体酸2;最后,在无水二氯甲烷中,在1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐存在下将中间体酸2用n-羟基琥珀酰亚胺活化转化为活化酯3,再在dmf中,在三乙胺存在下,用活化酯3分别与6-乙氨基-β-环糊精4、6-丁氨基-β-环糊精11或6-己氨基-β-环糊精13搅拌反应制备得到式(vi)、(xi)或(xii)所示的前药,记作cd-s-s-cpt-1、15和16。
本发明所述制备方法优选的另一种实施方式中,制备了式(vii)所示的前药cd-c-c-cpt,其合成路线如下:
具体包括如下步骤:
在4-(二甲基氨基)吡啶的存在下,在二氯甲烷中,使用三光气活化喜树碱的羟基,然后与过量的1,6-己二醇反应,制备得到中间体醇5;然后在无水二氯甲烷中在4-二甲氨基吡啶存在下使用丁二酸酐将中间体醇5转化为中间体酸6;最后,在无水二氯甲烷中,在1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐存在下将中间体酸6用n-羟基琥珀酰亚胺活化转化为活化酯7,再在dmf中,在三乙胺存在下,用活化酯7与6-乙氨基-β-环糊精4搅拌反应制备得到式(vii)所示的前药cd-c-c-cpt。
本发明所述制备方法优选的再一种实施方式中,制备了式(viii)~(x)和(xiii)所示的前药,其合成路线如下:
具体包括如下步骤:
将所述的中间体醇1溶于二甲亚砜中,分别加入羰基咪唑或炔丁酸,室温下搅拌反应得到活化酯9或17,所得的活化酯9或17溶于二甲亚砜中,加入6-乙氨基-β-环糊精4、6-丁氨基-β-环糊精11、6-己氨基-β-环糊精13或者6-叠氮-β-环糊精19,室温搅拌反应,可分别得到式(viii)~(x)或(xiii)所示的前药,记作10、13、14或20。
本发明的前药可以直接单独用作癌症治疗药物,也可以进一步制成其他药物学上可接受的剂型用于癌症治疗。因此,本发明还提供所述喜树碱类-环糊精前药在药物学上可接受的制剂。
本发明优选的方案中,所述的制剂是含有本发明所述的喜树碱类-环糊精前药的纳米颗粒制剂。
所述的纳米颗粒制剂既包括所述的喜树碱类-环糊精前药分子在水溶液中自组装形成的纳米颗粒,也包括所述的喜树碱类-环糊精前药分子与其他聚合物一起组装制备得到的纳米颗粒。
本发明还提供所述的喜树碱类-环糊精前药在制备癌症治疗药物中的应用。
本发明优选的一种应用方案中,使用高分子聚合物对本发明所述的喜树碱类-环糊精前药进行自组装,制备得到纳米颗粒。所述的高分子聚合物选自聚乙二醇-喜树碱、含有环糊精的嵌段聚合物或含有喜树碱的嵌段聚合物中的任意一种。
本发明的一种优选的实施方式中,将本发明所述的喜树碱类-环糊精前药与聚乙二醇-喜树碱或含有环糊精的嵌段聚合物以一定比例溶于水,搅拌或者超声一定时间制得纳米颗粒。
本发明的另一种优选的实施方式中,将本发明所述的喜树碱类-环糊精前药分子与含有喜树碱的嵌段聚合物以一定比例一起溶于水和丙酮的混合溶剂中,搅拌或者超声一定时间挥发掉有机溶剂,制得纳米颗粒。
本发明优选的另一种应用方案中,使用两亲性高分子对所述的喜树碱类-环糊精前药进行物理包裹,制备得到纳米颗粒。
本发明的一种优选的实施方式中,使用聚乙二醇-b-聚乳酸作为药用载体对所述的喜树碱类-环糊精前药进行物理包裹;具体是将本发明所述的喜树碱类-环糊精前药和聚乙二醇-b-聚乳酸以一定比例一起溶于有机溶剂(如四氢呋喃),然后滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒。
本发明的另一种优选的实施方式中,使用聚乙二醇-b-聚磷酯作为药用载体对所述的喜树碱类-环糊精前药进行物理包裹;具体是将本发明所述的喜树碱类-环糊精前药和聚乙二醇-b-聚磷酯以一定比例一起溶于有机溶剂(如四氢呋喃),然后滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒。
本发明的再一种优选的实施方式中,使用聚乙二醇-b-聚己内酯作为药用载体对所述的喜树碱类-环糊精前药进行物理包裹;具体是将本发明所述的喜树碱类-环糊精前药和聚乙二醇-b-聚己内酯以一定比例一起溶于有机溶剂(如四氢呋喃),然后滴入正在搅动或者超声的水溶液中,然后挥发掉有机溶剂,制得纳米颗粒。
在本发明中,我们创新性地把喜树碱类连接在环糊精上,制备出一种新颖的超分子化疗药物。这一药物在水溶液中可以通过环糊精与喜树碱之间的输水相互作用力自组装形成纳米粒子。所述的纳米粒子可以有效被肿瘤细胞摄取,并且具有载药效率高、载药量大的优点,而且喜树碱包结在环糊精的输水空腔中,可以大大提高喜树碱的水溶性,同时有效抑制喜树碱自身的水解,这一特性可以大大提高这一超分子药物的抗肿瘤活性。更重要的是喜树碱和环糊精之间的二硫键可以在肿瘤细胞内谷胱甘肽(gsh)的作用下断开释放出具有高抗癌活性的未经修饰的喜树碱,实现降低对正常组织毒性的同时提高抗肿瘤活性。截止目前,这一新颖的设计还未被国内外报道,具有非常高的创新价值。
本发明合成的喜树碱类-环糊精超分子化疗药物cd-s-s-cpt具有如下特点:
(1)有效提高喜树碱在生理环境中的溶解度和分散性;
(2)有效提高喜树碱在生理环境中的稳定性,防止水解;
(3)喜树碱被化学修饰后,在二硫键断裂前对细胞的毒性小;
(4)由于喜树碱和环糊精用二硫键连接,因此该前药对细胞内的还原性谷胱甘肽敏感,接触谷胱甘肽后能迅速释放喜树碱;
(5)本发明合成的超分子药物也可以很容易的被生物相容性高分子包裹制备出其他高载药量的纳米药物;
(6)最后,前药的合成简单,可以大批量生产,纳米颗粒制备容易,非常有潜力用于临床转化。
附图说明
图1为实施例1制备的cpt-s-s-oh的1hnmr谱图(300mhz,cd2cl2)。
图2为实施例2制备的cpt-s-s-cooh的1hnmr谱图(300mhz,cdcl3)。
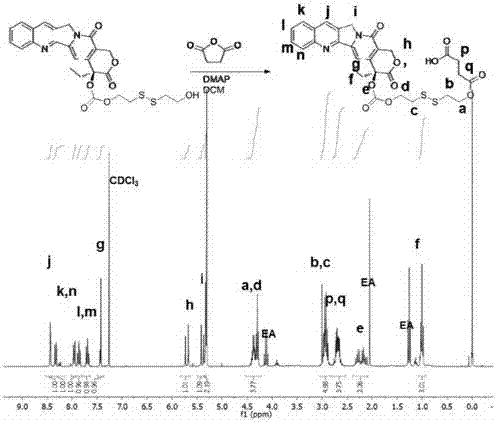
图3为实施例3制备的cd-s-s-cpt的1hnmr谱图(300mhz,dmso-d6)。
图4为实施例4制备的cpt-c-c-oh1hnmr谱图(300mhz,cd3cl)。
图5为实施例5制备的cpt-c-c-cooh1hnmr谱图(300mhz,cdcl3)。
图6为超分子前药cd-s-s-cpt制备得到的纳米颗粒的透射电镜照片。
图7为超分子前药cd-s-s-cpt与甲基聚乙二醇-喜树碱制备得到的纳米颗粒的透射电镜照片。
图8为药物从实施例3的cd-s-s-cpt纳米颗粒随时间释放的曲线图。
图9为实施例3的前药cd-s-s-cpt和喜树碱的随时间水解的曲线图。
图10为细胞毒性对比实验结果曲线图。
具体实施方式
下面通过实施例对本发明进行具体描述,本实施例只用于对本发明作进一步的说明,不能理解为对本发明保护范围的限制,本领域的技术人员根据上述发明的内容做出的一些非本质的改进和调整,均属本发明保护范围。
实施例1:cpt-s-s-oh(1)的制备
室温搅拌下,将4-二甲氨基吡啶(2.10g,17.2mmol)加入到喜树碱(2.00g,5.74mmol)的二氯甲烷溶液中。继续搅拌30分钟后,缓慢加入三光气(0.633g,2.12mmol)。搅拌1小时后,逐滴加入2,2'-二硫代二乙醇(17.2g,117mmol),反应液室温搅拌过夜。将混合物用饱和食盐水(100ml)洗涤三次,分液得到的二氯甲烷有机相用无水硫酸钠干燥。使用预填充的二氧化硅柱通过快速色谱分离纯化产物。产量:2.12g(产率为70%)。1hnmr(300mhz,cdcl3):8.46(s,1h),8.22(d,j=8.3hz,1h),7.99(dd,j=8.2,1.1hz,1h),7.86(ddd,j=6.9,6.5hz,1h),7.70(ddd,j=8.1,6.9,1.2hz,1h),7.42(s,1h),5.64(d,j=18hz,1h),5.36(m,1h)4.37(t,j=6.0hz,2h),3.93-3.81(m,2h),3.03-2.82(m,4h),2.19(tdd,j=14.0,12.3,7.5hz,3h)1.01(t,j=7.5hz,3h)。图1为cpt-ss-oh的1hnmr谱图,证明了制备该化合物成功。
实施例2:cpt-s-s-cooh(2)的制备
在搅拌下将实施例方法1制备的cpt-s-s-oh(300mg,0.552mmol)溶于无水二氯甲烷中,分别加入丁二酸酐(1.00g,10.0mmol)和少量4-二甲氨基吡啶(21.0mg,0.172mmol),反应液室温搅拌过夜。溶液选干后,用水(100ml)洗涤固体三次,真空干燥得到产物cpt-s-s-cooh(产率为88%)。1hnmr(300mhz,cdcl3):8.44(s,1h),8.32(d,j=8.4hz,1h),7.95(d,j=8.2hz,1h),7.84(ddd,j=8.5,6.9,1.4hz,1h),7.69(ddd,j=8.1,7.0,1.1hz,1h),7.43(s,1h),5.71(d,j=17.3hz,1h),5.39(d,j=17.3hz,1h),5.32(s,2h),4.46-4.25(m,4h),3.00-2.86(m,4h),2.79-2.61m,2h),1.01(t,j=7.5,3h)。esi-msm/z:计算值628.12,实测值629.10(m+h)+。图2为cpt-s-s-cooh的1hnmr谱图,证明了制备该化合物成功。
实施例3:cd-s-s-cpt的制备
将按实施例2方法制备的cpt-s-s-cooh(628mg,1.00mmol)溶于100ml无水二氯甲烷中。分别加入n-羟基琥珀酰亚胺(0.575g,5.00mmol),1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc.hcl,1.91g,10.0mmol),室温下搅拌1天。有机溶剂用旋转蒸发仪除去,混合物用冷冻的甲醇洗涤3次(100ml)。得到的白色固体(363mg,0.500mmol)溶于dmf中(20ml),分别加入6-乙氨基-β-环糊精(1.18g,1.00mmol)和三滴三乙胺,室温搅拌过夜。溶液倒入100丙酮中,抽滤得到浅黄色固体,二氯甲烷洗涤三次(100ml),水洗三次(50ml),固体抽干得到产品cd-s-s-cpt(产率74%)。1hnmr(300mhz,cdcl3):8.45(s,1h),8.33(d,j=8.4hz,1h),7.94(d,j=8.2hz,1h),7.85(ddd,j=8.5,6.9,1.4hz,1h),7.71(ddd,j=8.1,7.0,1.1hz,1h),7.43(s,1h),5.71(d,j=6hz,1h),5.39(d,j=6hz,1h),5.32(s,2h),4.96-4.88(m,7h),4.46-4.25(m,4h),3.83-3.68(m,28h),3.51-3.25(m,14h),3.00-2.86(m,4h),2.89(t,j=6hz,2h),2.79-2.61m,2h),1.01(t,j=7.5,3h)。esi-msm/z:计算值1787.72,实测值1788.86(m+h)+。图3为cd-s-s-cpt的1hnmr谱图,证明了制备该化合物成功。
实施例4:cd-c-c-oh(5)的制备
室温搅拌下,将4-二甲氨基吡啶(2.10g,17.2mmol)加入到喜树碱(2.00g,5.74mmol)的二氯甲烷溶液中。继续搅拌30分钟后,缓慢加入三光气(0.633g,2.12mmol)。搅拌1小时后,逐滴加入1,6-己二醇(11.8g,100mmol),反应液室温搅拌过夜。将混合物用饱和食盐水(100ml)洗涤三次,分液得到的二氯甲烷有机相用无水硫酸钠干燥。使用预填充的二氧化硅柱通过快速色谱分离纯化产物。产量:1.78g(产率为63%)。1hnmr(300mhz,cdcl3):8.41(s,1h),8.23(d,j=8.7hz,1h),7.95(dd,j=8.2,1.1hz,1h),7.85(ddd,j=6.9,6.5hz,1h),7.68(ddd,j=8.1,6.9,1.2hz,1h),7.36(s,1h),5.70(d,j=17.3hz,1h),5.39(d,j=1h),5.30(s,2h),4.21-4.05(m,2h),3.59(dd,j=10.1,6.1hz,2h),2.43-2.01(m,2h),1.74-1.65(m,3h),1.59-1.47(m,2h),1.45-1.32(m,4h),1.00(t,j=7.5hz,3h)。图4为cpt-c-c-oh的1hnmr谱图,证明了制备该化合物成功。
实施例5:cpt-c-c-cooh(6)的制备
在搅拌下将按实施例4方法制备的cpt-c-c-oh(492mg,1.00mmol)溶于无水二氯甲烷中,分别加入丁二酸酐(1.00g,10.0mmol)和少量4-二甲氨基吡啶(21.0mg,0.172mmol),反应液室温搅拌过夜。溶液选干后,用水(100ml)洗涤固体三次,真空干燥得到产物cpt-c-c-cooh。产量485mg(产率为82%)。esi-msm/z:计算值592,实测值591(m-h)-。1hnmr(300mhz,cdcl3):8.42(s,1h),8.28(d,j=8.7hz,1h),7.95(d,j=8.3hz,1h),7.85(ddd,j=1h),7.68(ddd,j=8.1,6.9,1.1hz,1h),7.38(d,j=5.1hz,1h),5.70(d,j=17.3hz,1h),5.39(d,j=17.3hz,1h),5.31(s,2h),4.23-3.99(m,4h),2.74-2.56(m,4h),2.37-2.07(m,2h),1.61(ddd,j=12.9,6.6hz,5h),1.44-1.23(m,7h),1.00(t,j=7.5hz,3h)。图5为cpt-c-c-cooh的1hnmr谱图,证明了制备该化合物成功。
实施例6:cd-c-c-cpt的制备
将按实施例5方法制备得到的cpt-c-c-cooh(1.18g,2.00mmol)溶于200ml无水二氯甲烷中。分别加入n-羟基琥珀酰亚胺(1.15g,10.0mmol),1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc.hcl,3.82g,20.0mmol),室温下搅拌1天。有机溶剂用旋转蒸发仪除去,混合物用冷冻的甲醇洗涤3次(每次用量100ml)。得到的白色固体溶于dmf中(40ml),分别加入6-乙氨基-β-环糊精(1.18g,1.00mmol)和三滴三乙胺,室温搅拌过夜。溶液倒入100丙酮中,抽滤得到浅黄色固体,二氯甲烷洗涤三次(每次100ml),水洗三次(每次20ml),固体抽干得到产品cd-c-c-cpt(产率68%)。1hnmr(300mhz,cdcl3):8.43(s,1h),8.30(d,j=8.7hz,1h),7.93(d,j=8.3hz,1h),7.84(ddd,j=1h),7.66(ddd,j=8.1,6.9,1.1hz,1h),7.39(d,j=5.1hz,1h),5.71(d,j=17.3hz,1h),5.40(d,j=17.3hz,1h),5.32(s,2h),4.92(m,7h),4.21-4.00(m,4h),3.85–3.64(m,28h),3.53–3.21(m,14h),2.86(t,j=6hz,2h),2.76-2.53(m,4h),2.39-2.05(m,2h),1.62(ddd,j=12.9,6.6hz,5h),1.45-1.22(m,7h),1.01(t,j=7.5hz,3h)。esi-msm/z:计算值1751.67,实测值1752.73(m+h)+,证明了制备该化合物成功。
实施例7:前药分子10,13和14的制备
将按实施例1方法制备得到的cpt-s-s-oh(528mg,1.00mmol)溶于50ml二甲亚砜中。分别加入羰基咪唑(1.62g,10.0mmol),室温下搅拌1天。反应液倒入冰甲醇中,得到的白色沉淀用乙醚洗涤三次(每次50ml)。所得的产品溶于二甲亚砜中(40ml),加入6-乙氨基-β-环糊精(1.18g,1.00mmol),室温搅拌过夜。溶液倒入100丙酮中,抽滤得到浅黄色固体,二氯甲烷洗涤三次(每次100ml),水洗三次(每次10ml),固体抽干得到产品10(产率57%)。1hnmr(300mhz,cdcl3):8.43(s,1h),8.32(d,j=8.4hz,1h),7.91(d,j=8.2hz,1h),7.83(ddd,j=8.5,6.9,1.4hz,1h),7.70(ddd,j=8.1,7.0,1.1hz,1h),7.45(s,1h),5.70(d,j=6hz,1h),5.38(d,j=6hz,1h),5.34(s,2h),4.96-4.88(m,7h),4.46-4.25(m,4h),3.83-3.68(m,28h),3.51-3.25(m,14h),2.89(t,j=6hz,2h),2.79-2.61m,2h),1.01(t,j=7.5,3h)。esi-msm/z:计算值1731.66,实测值1732.77(m+h)+,证明了制备该化合物成功。
同样的方法下,我们分别采用6-丁氨基-β-环糊精或6-己氨基-β-环糊精替代所述的6-乙氨基-β-环糊精后,可以分别制备出另外两种超分子前药13和14。
化合物13:1hnmr(300mhz,cdcl3):8.43(s,1h),8.32(d,j=8.4hz,1h),7.91(d,j=8.2hz,1h),7.83(ddd,j=8.5,6.9,1.4hz,1h),7.70(ddd,j=8.1,7.0,1.1hz,1h),7.45(s,1h),5.70(d,j=6hz,1h),5.38(d,j=6hz,1h),5.34(s,2h),4.96-4.88(m,7h),4.46-4.25(m,4h),3.82-3.66(m,28h),3.50-3.27(m,14h),2.87(t,j=6hz,2h),2.79-2.61m,2h),1.52(m,2h),1.38(m,2h),1.01(t,j=7.5,3h)。esi-msm/z:计算值1759.71,实测值1760.80(m+h)+。
化合物14:1hnmr(300mhz,cdcl3):8.43(s,1h),8.32(d,j=8.4hz,1h),7.91(d,j=8.2hz,1h),7.83(ddd,j=8.5,6.9,1.4hz,1h),7.70(ddd,j=8.1,7.0,1.1hz,1h),7.45(s,1h),5.70(d,j=6hz,1h),5.38(d,j=6hz,1h),5.34(s,2h),4.96-4.88(m,7h),4.46-4.25(m,4h),3.82-3.66(m,28h),3.50-3.27(m,14h),2.87(t,j=6hz,2h),2.79-2.61m,2h),1.50(m,2h),1.36(m,2h),1.27(m,4h),1.01(t,j=7.5,3h)。esi-msm/z:计算值1787.77,实测值1788.79(m+h)+。证明了制备该化合物成功。
实施例8:前药分子15和16的制备
将按实施例2方法制备的cpt-s-s-cooh(628mg,1.00mmol)溶于100ml无水二氯甲烷中。分别加入n-羟基琥珀酰亚胺(0.575g,5.00mmol),1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc.hcl,1.91g,10.0mmol),室温下搅拌1天。有机溶剂用旋转蒸发仪除去,混合物用冷冻的甲醇洗涤3次(100ml)。得到的白色固体(363mg,0.500mmol)溶于dmf中(20ml),分别加入6-丁氨基-β-环糊精(2.41g,2.00mmol)和三滴三乙胺,室温搅拌过夜。溶液倒入100丙酮中,抽滤得到浅黄色固体,二氯甲烷洗涤三次(100ml),水洗三次(50ml),固体抽干得到产品15(产率66%)。1hnmr(300mhz,cdcl3):8.44(s,1h),8.35(d,j=8.4hz,1h),7.94(d,j=8.2hz,1h),7.85(ddd,j=8.5,6.9,1.4hz,1h),7.71(ddd,j=8.1,7.0,1.1hz,1h),7.43(s,1h),5.71(d,j=6hz,1h),5.40(d,j=6hz,1h),5.32(s,2h),4.96-4.88(m,7h),4.46-4.25(m,4h),3.83-3.68(m,28h),3.51-3.25(m,14h),3.00-2.86(m,4h),2.89(t,j=6hz,2h),2.79-2.61m,2h),1.52(m,2h),1.38(m,2h),1.01(t,j=7.5,3h)。esi-msm/z:计算值1815.78,实测值1816.81(m+h)+,证明了制备该化合物成功。
同样的方法下,采用6-己氨基-β-环糊精替代6-丁氨基-β-环糊精后,我们可以制备出化合物16(产率63%)。1hnmr(300mhz,cdcl3):8.46(s,1h),8.33(d,j=8.4hz,1h),7.96(d,j=8.2hz,1h),7.84(ddd,j=8.5,6.9,1.4hz,1h),7.71(ddd,j=8.1,7.0,1.1hz,1h),7.44(s,1h),5.71(d,j=6hz,1h),5.38(d,j=6hz,1h),5.32(s,2h),4.96-4.88(m,7h),4.46-4.25(m,4h),3.83-3.68(m,28h),3.51-3.25(m,14h),3.00-2.86(m,4h),2.89(t,j=6hz,2h),2.79-2.61(m,2h),1.54(m,2h),1.37(m,2h),1.26(m,4h),1.01(t,j=7.5,3h)。esi-msm/z:计算值1843.83,实测值1844.90(m+h)+,证明了制备该化合物成功。
实施例9:前药分子20的制备
将按实施例1方法制备的cpt-s-s-oh(628mg,1.00mmol)溶于100ml无水二氯甲烷中。分别加入炔丁基酸(840mg,10.0mmol),1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐(edc.hcl,3.82g,20.0mmol),室温下搅拌1天。有机溶剂用旋转蒸发仪除去,混合物用冷冻的甲醇洗涤3次(100ml)。得到的白色固体溶于dmf中(20ml),分别加入6-叠氮-β-环糊精(2.32g,2.00mmol),cuso4·5h2o,(24.9mg,0.100mmol),抗坏血酸钠(198mg,1.00mmol),鼓氮气30分钟,在氮气保护下室温搅拌过夜。溶液倒入100丙酮中,抽滤得到浅黄色固体,二氯甲烷洗涤三次(100ml),水洗三次(50ml),固体抽干得到产品20(产率71%)。esi-msm/z:计算值1754.65,实测值1755.71(m+h)+,证明了制备该化合物成功。
实施例10:超分子前药cd-s-s-cpt制备纳米颗粒
取实施例3方法制备的cd-s-s-cpt(2.00mg)溶于磷酸缓冲液中(pbs,4ml,ph=7.4),超声20分钟后得到澄清溶液。从透射电镜照片(图6)中可以看出cd-s-s-cpt可以自组装形成直径约为40nm的纳米粒子。这些纳米粒子的稳定性非常高,可以在溶液中保存三个月不发生任何沉淀。同样,按照实施例6-9的方法制备的一系列其他超分子前药也可以自组装后形成高稳定性的纳米粒子。
另外,本发明的方法制备的一系列超分子前药还可以与聚合物混合使用,制备出聚合物纳米颗粒。举例来说,取实施例3制备的cd-s-s-cpt(5.00mg),甲基聚乙二醇-喜树碱(25.0mg)溶于磷酸缓冲液中(pbs,12ml,ph=7.4),超声30分钟后得到澄清溶液。从透射电镜照片(图7)中可以看出所得的纳米粒子直径约为80nm。这些纳米粒子的稳定性非常高,可以在溶液中保存三个月不发生任何沉淀。同样的,cd-s-s-cpt等本发明的一系列前药分子可以与含有环糊精的嵌段聚合物或者含有喜树碱的嵌段聚合物通过主客体相互作用制备出粒径约为100nm的纳米粒子。这些纳米粒子具有很高的稳定性,可以长时间保存在pbs溶液中不发生沉淀。
实施例11:药物释放实验
通过透析法测量喜树碱的释放。简言之,用磷酸盐缓冲盐水(pbs)将实施例3方法制备的cd-s-s-cpt配制一定浓度的纳米粒子溶液(1.00mg/ml),然后将5ml的cd-s-s-cpt溶液转移到预浸渍的透析袋(mwco:1kda)中,并分别用pbs溶液或者含有不同浓度的gsh溶液(1.00mm,5.00mm,10.0mm)透析48小时。通过hplc(条件:30%乙腈,含0.1%tfa,1ml/min,uv检测器,250nm)监测透析袋中cpt的浓度,绘制出不同条件下喜树碱药物分子的释放曲线。每个实验重复3次。
结果发现(图8)不加gsh的条件下喜树碱释放非常缓慢。在gsh的存在下,喜树碱释放显著加快。另一方面,随着gsh浓度的提高,喜树碱释放速度也变快。这一结果证明cd-s-s-cpt可以在gsh的存在下,通过gsh与双硫键反应释放出喜树碱。
同样,按照实施例7-9的方法制备的一系列其他超分子前药也与上述cd-s-s-cpt一样,制备成纳米粒子溶液后,在gsh的存在下,具有与cd-s-s-cpt类似的喜树碱释放效果。
实施例12:cd-s-s-cpt和喜树碱的水解实验
用20ml二甲亚砜(dmso)和ph为7.4的pbs缓冲液的混合溶液(体积比为1:1)分别溶解喜树碱和实施例3方法制备的cd-s-s-cpt(喜树碱浓度保持在1mg/ml)配成溶液,室温搅拌。每隔一定时间,通过hplc(条件:30%乙腈,含0.1%tfa,1ml/min,uv检测器,250nm)监测喜树碱溶液和cd-s-s-cpt溶液中喜树碱分子内酯结构的百分数,绘制出不同时间下喜树碱分子的水解曲线(图9)。每个实验重复3次。
结果显示(图9)在环糊精包结下cd-s-s-cpt中喜树碱水解大大下降,证明了这一超分子药物相比较于喜树碱本身可以有效抑制药物水解,有利于提高其药效;本发明的其他一系列前药分子也基于相同的原理能有效抑制药物水解。
实施例:13:体外细胞毒性测试
将人结肠癌细胞hct116接种在96孔板的mccoy's5a培养基(10%胎牛血清和1%青霉素)中。将细胞在37℃下在含有5%co2的潮湿气氛中孵育。接种后24小时用新鲜培养基替换培养基。测试制剂选择喜树碱、cd-s-s-cpt和cd-c-c-cpt,将每种制剂溶解于pbs中并使用细胞培养基稀释。对于每个孔,加入100μl具有不同指定药物浓度的细胞培养基。通过加入100μl培养基产生阴性对照。将细胞孵育48小时后,用含有0.5mg/ml3-(4,5-二甲基噻唑-2-基)-2,5-二苯基四氮唑(mtt)的100μl培养基替换培养基。孵育4小时后,除去含有mtt的培养基,用pbs小心洗涤3次。然后,加入dmso(100μl),在bioteksynergyh4读数器中测量在570nm的波长下吸光度。使用graphpadprism5进行ic50值的计算和统计分析。
如图10显示,喜树碱的ic50为0.14μm,cd-s-s-cpt为0.17μm,两者对hct116细胞都具有很好的细胞毒性。证明本发明的超分子化疗药物中,cd-s-s-cpt的喜树碱和环糊精具有很强的抗癌活性;cd-c-c-cpt的ic50为1.92μm,表明含有二硫键的前药分子细胞毒性比非二硫键的高很多,说明二硫键断裂并且释放喜树碱是维持药物毒性的关键所在。基于相同的原理,本发明的其他一系列二硫键连接的前药分子也具有很强的抗癌活性。
- 还没有人留言评论。精彩留言会获得点赞!