1‑(3‑N‑取代‑咔唑基)‑3‑芳基‑3‑(2‑环己酮基)‑丙酮及其制备方法与流程
本发明属于化学合成领域,特别涉及一种1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮及其制备方法。
背景技术:
咔唑是一种重要的杂环化合物,首先,它本身所含有的共轭体系较大且分子内强的电子转移,具有的给电子能力和空穴传输能力较强,及热稳定性和光化学稳定性较高;其次,咔唑化合物的衍生物在光电材料、染料、医药、超分子识别、化学传感等领域具有潜在的广泛应用。其生物活性和药用意义引起了广大化学工作者的研究兴趣。氮唑类化合物因在医药(抗菌、抗癌、抗阻胺剂、抗氧化、抗炎、神经抑制等)、染料、材料等多领域广阔的应用前景一直备受研究学者的青睐。由此可见,咔唑类化合物的研究成为最有吸引力的研究目标。
1,3-二苯基丙烯酮是由芳香醛酮经过缩合形成的,又被称作查尔酮。它在医药领域的主要作用是抗炎、抗菌,在农药领域的主要作用是有良好的除草功效,同时也是合成黄酮类化合物的重要中间体,因此在医药和农药领域被广泛应用。查尔酮化合物在适当催化条件下可打开其中的碳碳双键从而发生一系列的加成反应,比如micheal加成反应,因此,查尔酮在有机合成中的应用前景十分广泛。
在有机合成化学中,碳负离子与α,β-不饱和共轭体系(醛、酮、酯、腈和硝基化合物等)进行的共轭加成反应叫做michael(麦克尔)加成反应。目前关于含咔唑基的michael加成产物的相关报道非常少,且大都是采用强碱做催化剂,强碱容易腐蚀设备,造成成本加大。
技术实现要素:
本发明的目的在于提供一种1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮及其制备方法,该方法操作简单、反应条件温和,制得的产品具有杀菌、抑菌、除草等功效。
为达到上述目的,本发明采用的技术方案为:
一种1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮,其结构通式如下:
其中,ar为苯基、卤代苯基、甲基苯基、甲氧基苯基、硝基苯基、羟基苯基、氨基苯基、乙烯基苯基或五元杂环基;
r为甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、苯氧亚甲基、对氯苯氧亚甲基、邻氯苯氧亚甲基、2,4-二氯苯氧亚甲基、间氯苯氧亚甲基、对氟苯氧亚甲基、对溴苯氧亚甲基、对碘苯氧亚甲基、对甲氧基苯氧亚甲基、2-硝基苯氧亚甲基、苯氧乙基、α-萘氧亚甲基、β-萘氧亚甲基或β-萘氧乙基。
所述的卤代苯基为邻氟苯基、对氟苯基、邻氯苯基、对氯苯基、2,4-二氯苯基、邻溴苯基、间溴苯基或对溴苯基;
所述的甲基苯基为邻甲基苯基、间甲基苯基或对甲基苯基;
所述的甲氧基苯基为间甲氧基苯基或对甲氧基苯基;
所述的硝基苯基为间硝基苯基、3,5-二硝基苯基或对硝基苯基;
所述的羟基苯基为邻羟基苯基或对羟基苯基;
所述的氨基苯基为邻氨基苯基、间氨基苯基或对氨基苯基;
所述的五元杂环基为呋喃基或噻吩基。
所述的1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮的制备方法,包括以下步骤:
步骤1)将amol1-(3-n-取代-咔唑基)-3-芳基-丙烯酮、bmol碱性催化剂、cmol环己酮和溶剂无水乙醇加入反应容器中,进行加热回流反应,其中a:b:c=1:(5~6):(5~6);
步骤2)反应完毕后,将反应液冷却至室温,加入0~5℃的冷水,有固体析出,对析出的固体进行水洗、抽滤,滤饼重结晶,然后进行干燥,即得到1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮。
所述1-(3-n-取代-咔唑基)-3-芳基-丙烯酮的制备方法为:以乙酰基咔唑、芳香醛为原料,以naoh/k2co3为催化剂进行研磨反应,反应完毕后反复水洗、干燥得到粗品,粗品经硅胶柱层析法进行分离提纯,即得到1-(3-n-取代-咔唑基)-3-芳基-丙烯酮。
所述加热回流的反应温度为70~80℃,反应时间为0.5~1.5h。
所述加热回流反应过程中用tlc监测反应进程,当1-(3-n-取代-咔唑基)-3-芳基-丙烯酮的原料点消失时表示原料反应完全;其中tlc的展开剂是体积比为1:3的乙酸乙酯与石油醚的混合溶剂。
所述步骤1)中加入dml无水乙醇,a:d=1:(20000~30000)。
所述加热回流反应过程中以60~80rad/min的速度对反应体系进行搅拌。
所述碱性催化剂为粒状的固体naoh。
所述步骤2)中对析出的固体反复进行水洗、抽滤,直到滤液的ph值呈中性,然后用无水乙醇对滤饼进行重结晶,再在20~30℃下干燥20~30h。
相对于现有技术,本发明的有益效果为:
本发明探索了含新的取代基团的咔唑基michael加成产物以及制备方法,并优化了实验条件,缩短了反应时间,为绿色合成做出了贡献。本发明提供的1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮,是一系列全新的酮类化合物,具有杀菌、抑菌、除草等功效,可用于医药领域和农药领域等领域。
本发明提供的1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮的制备方法,将1-(3-n-取代-咔唑基)-3-芳基-丙烯酮、碱性催化剂、环己酮和溶剂无水乙醇加入反应容器中加热回流反应,对反应产物进行后处理即得到目标产物。与传统合成方法相比,本发明的反应现象明显,反应过程简单,操作简单,反应时间短,反应条件温和,水浴下即可进行反应,设备要求低,且该方法的后处理简单,副产物少,产物的产率高(高达94%)。克服了传统合成方法设备要求高,反应时间长等缺点,具有经济、方便、高效、绿色的优点。
进一步的,本发明在制备过程中,用tlc监测反应过程,所用的展开剂为体积比为1:3的乙酸乙酯和石油醚,监测准确,利于控制反应进度和结束。同时本发明所用的催化剂为粒状的固体naoh,廉价易得,催化效果良好,使反应更加迅速和完全,且后处理简单。
附图说明
图1为实施例1制得的产物的红外图谱;
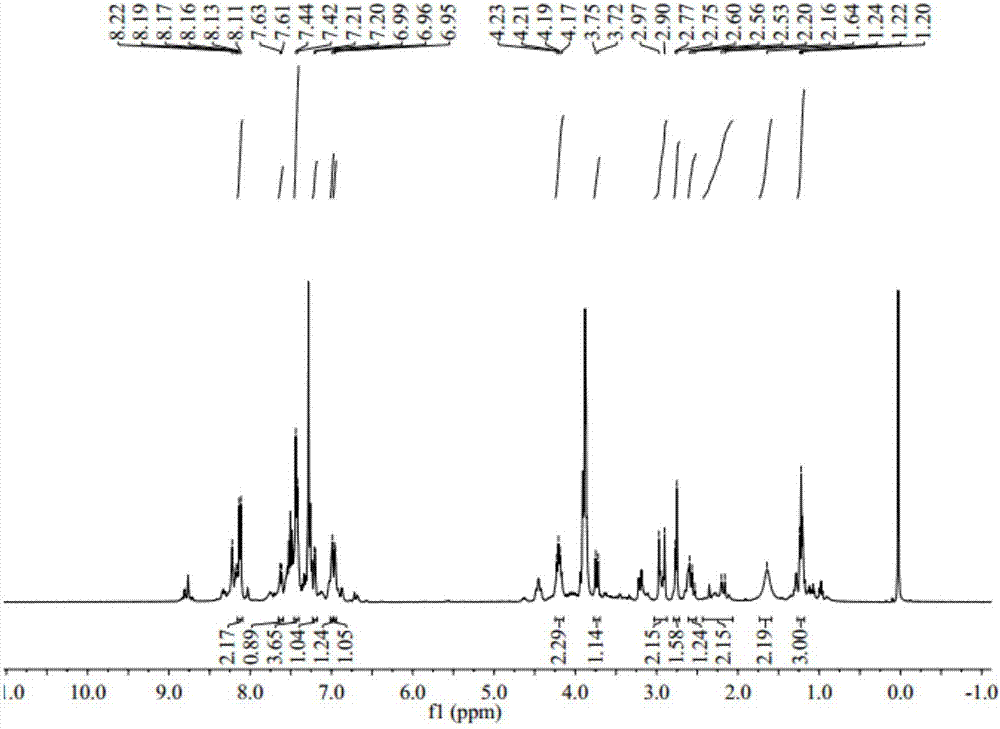
图2为实施例2制得的产物的1hnmr图谱;
图3为实施例3制得的产物的红外图谱;
图4为实施例6制得的产物的1hnmr图谱。
具体实施方式
本发明提供的1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮的反应方程式如下:
其中ar为芳基,具体为苯基、对氯苯基、对溴苯基、对氟苯基、对甲基苯基、对甲氧基苯基、间硝基苯基、对羟基苯基、邻羟基苯基、乙烯基苯基、邻甲基苯基、间甲基苯基、间甲氧基苯基、2-氟苯基、对氨基苯基、间氨基苯基、邻氨基苯基、邻氯苯基、2,4-二氯苯基、对硝基苯基、3,5-二硝基苯基、邻溴苯基、间溴苯基、呋喃基或噻吩基。
r为甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基、壬基、苯氧亚甲基、对氯苯氧亚甲基、邻氯苯氧亚甲基、2,4-二氯苯氧亚甲基、间氯苯氧亚甲基、对氟苯氧亚甲基、对溴苯氧亚甲基、对碘苯氧亚甲基、对甲氧基苯氧亚甲基、2-硝基苯氧亚甲基、苯氧乙基、α-萘氧亚甲基、β-萘氧亚甲基或β-萘氧乙基。
下面将结合本发明较佳的实施例对本发明做进一步详细说明。
实施例1
依次称取0.006mol环己酮,0.006molnaoh(固体、粒状),30ml无水乙醇,加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,待混合均匀后,再加入0.001mol的1-(3-n-乙基-咔唑基)-3-(对甲氧基苯基)-丙烯酮,搅拌并缓慢升温至80℃,使其在80℃的温度及的70rad/min搅拌速度下回流反应40min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml3℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在25℃真空干燥25h,得到粉末状产品,即为1-(3-n-乙基-咔唑基)-3-(对甲氧基苯基)-3-(2-环己酮基)-丙酮。m.p.154℃-158℃。
ir(kbr,v/cm-1):2935,1369(-ch3),2838,1494(-ch2),3053,1626,1584,1508,1446(ar-h),1709,1667(c=o),1327(-nr2).
由图1可见,2935、1369cm-1分别为-ch3的伸缩振动吸收峰和弯曲振动峰,2838、1494cm-1为-ch2的伸缩振动吸收峰和弯曲振动峰,1709、1667cm-1为c=o的伸缩振动峰,1327cm-1为n-r2的伸缩振动峰,1626、1584、1508、1446cm-1为苯环的骨架振动吸收特征峰,3053cm-1为芳环中不饱和h的特征吸收峰。
1hnmr:4.09-4.14(q,2h,n-ch2-),1.08-1.13(t,3h,-ch3),3.77(s,3h,-och3),2.35-2.47(d,2h,-coch2-),2.32-2.37(m,1h,-ch),3.42-3.55(m,1h,-ch),2.16-2.22(m,4h,-ch2),1.52-1.63(m,4h,-ch2),6.745-6.82(d,1h,thy-h),7.53-7.63(d,1h,thy-h),7.71-7.80(m,1h,thy-h),7.33-7.41(d,4h,ph-h),8.08-8.18(m,2h,ph-h),6.76(s,1h,ph-h).
实施例2
依次称取0.006mol环己酮,0.006molnaoh(固体、粒状),30ml无水乙醇,加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,待混合均匀后,再加入0.001mol1-(3-n-乙基-咔唑基)-3-(2-噻吩基)-丙烯酮,搅拌并缓慢升温至70℃,使其在70℃的温度及的70rad/min搅拌速度下回流反应30min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml3℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在25℃真空干燥25h,得到粉末状产品,即为1-(3-n-乙基-咔唑基)-3-(2-噻吩基)-3-(2-环己酮基)-丙酮。m.p.122℃-125℃。
ir(kbr,ν/cm-1):2923,1374(-ch3),1658,1628(c=o),1568,1414,1384(ar),1348(-nr2).
1hnmr:4.17-4.23(q,2h,n-ch2-),1.20-1.24(t,3h,-ch3),2.75-2.77(d,2h,-coch2-),2.53-2.60(m,1h,-ch),3.72-3.75(m,1h,-ch),2.16-2.20(m,4h,-ch2),1.64-1.68(m,4h,-ch2),6.95-6.96(d,1h,thy-h),7.20-7.21(d,1h,thy-h),7.63-7.61(m,1h,thy-h),7.42-7.44(d,4h,ph-h),8.13-8.22(m,2h,ph-h),6.99(s,1h,ph-h).
如图2所示,1.20-1.24ppm为甲基的质子峰,为三重峰,积分面积为3个h;4.23-4.17ppm处为咔唑基团上与n相连的亚甲基的质子峰,积分面积显示为2个h;2.75-2.77ppm处的双重峰为-coch2的质子峰;2.16-2.20ppm处和1.64-1.68ppm处出现了多重峰,分别代表了环己酮基团上的质子峰;2.53-2.60ppm和3.72-3.75ppm处的多重峰为与环己酮基相连的c上的质子峰和与噻吩基团相连的c上的质子峰,积分面积分别都是1个h;6.95-6.96ppm和7.20-7.21ppm处为双重峰,是噻吩基团上的质子峰,7.42-7.44ppm处的双重峰、8.13-8.22ppm处的多重峰、以及6.99ppm处的单峰代表的是咔唑基团中苯环中的质子峰。
实施例3
用称量天平称取0.006mol环己酮,0.006molnaoh(固体、粒状),30ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-甲基-咔唑基)-3-(对甲基苯基)-丙烯酮,搅拌并缓慢升温至80℃,使其在80℃的温度及的70rad/min搅拌速度下回流反应75min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml3℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在25℃真空干燥25h,得到咖啡色固体,即为1-(3-n-甲基-咔唑基)-3-(对甲基苯基)-3-(2-环己酮基)-丙酮。m.p.131℃-134℃。其ir谱图如图3所示。
ir:2935,1362(-ch3),2852,1480(-ch2),1709,1666(c=o),3046,1584,1432(ar-h),1327(nr2).
1hnmr:1.61-1.83(m,8h,-ch2),3.86(m,3h,-ch3),3.30-3.37(m,1h,-ch),2.75-2.77(d,2h,-ch2),2.57-2.63(m,1h,-ch),2.43(s,3h,-ch3),7.32-7.42(m,4h,ph-h),7.51-7.55(d,2h,ph-h),8.16(s,1h,ph-h),7.12-7.14(d,2h,ph-h),7.07-7.09(d,2h,ph-h).
实施例4
用称量天平称取0.006mol环己酮,0.006molnaoh(固体、粒状),30ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-乙基-咔唑基)-3-(对氯苯基)-丙烯酮,搅拌并缓慢升温至80℃,使其在80℃的温度及的70rad/min搅拌速度下回流反应70min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml1℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在25℃真空干燥25h,得到深棕色固体,即为1-(3-n-乙基-咔唑基)-3-(对氯苯基)-3-(2-环己酮基)-丙酮。m.p.100℃-102℃。
ir:2935,1369(-ch3),2859,1432(-ch2),1597,1479(ar),1722,1667(c=o),1321(nr2).
1hnmr:1.49-1.52(t,3h,-ch3),3.76-3.89(q,2h,-ch2),1.62-1.67(m,2h,-ch2),1.80-1.86(m,4h,-ch2),2.21-2.30(t,2h,-ch2),2.53-2.57(m,1h,-ch),2.74-2.79(m,2h,-coch2),3.53-3.59(m,1h,-ch),7.03-7.11(m,2h,ph-h),7.15-7.27(m,4h,ph-h),7.35-7.37(d,2h,ph-h),7.84-7.85(d,2h,ph-h)7.99(s,1h,ph-h).
实施例5
用称量天平称取0.006mol环己酮,0.006molnaoh(固体、粒状),30ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-乙基-咔唑基)-3-(对氟苯基)-丙烯酮,搅拌并缓慢升温至80℃,使其在80℃的温度及的70rad/min搅拌速度下回流反应70min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml3℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在25℃真空干燥25h,得到红棕色固体,即为1-(3-n-乙基-咔唑基)-3-(对氟苯基)-3-(2-环己酮基)-丙酮。m.p.112℃-115℃。
ir:2935,1370(-ch3),2852,1446(-ch2),1598,1480(ar),1702,1698(c=o),1321(nr2).
1hnmr:1.53-1.59(t,3h,-ch3),3.85(m,2h,-ch2),2.81-2.85(d,2h,-coch2),3.54-3.58(m,1h,-ch),2.41-2.50(m,1h,-ch),1.68-1.84(m,8h,-ch2),7.07-7.19(d,8h,ph-h),7.23-7.35(m,2h,ph-h),7.79(s,1h,ph-h).
实施例6
用称量天平称取0.006mol环己酮,0.006molnaoh(固体、粒状),30ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-甲基-咔唑基)-3-(对氟苯基)-丙烯酮,搅拌并缓慢升温至80℃,使其在80℃的温度及的70rad/min搅拌速度下回流反应80min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml3℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在25℃真空干燥25h,得到红棕色固体,即为1-(3-n-甲基-咔唑基)-3-(对氟苯基)-3-(2-环己酮基)-丙酮。m.p.114℃-116℃。
ir:2904,1369(-ch3),1598,1521,1480(ar),1713,1672(c=o),1335(nr2).
1hnmr:1.27-1.32(m,2h,-ch2),1.62-1.82(m,4h,-ch2),2.03-2.05(m,2h,-ch2),2.55-2.60(m,1h,-ch),2.75-2.77(d,2h,-coch2),3.63-3.68(q,1h,-ch),3.87(s,3h,-ch3),7.17-7.20(d,1h,ph-h),7.24-7.26(d,2h,ph-h),7.31-7.38(m,6h,ph-h),7.52-7.54(d,1h,ph-h),8.17(s,1h,ph-h).如图4所示。
实施例7
用称量天平称取0.005mol环己酮,0.005molnaoh(固体、粒状),20ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-丙基-咔唑基)-3-苯基-丙烯酮,搅拌并缓慢升温至74℃,使其在74℃的温度及的60rad/min搅拌速度下回流反应90min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml0℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在20℃真空干燥30h,即得到1-(3-n-丙基-咔唑基)-3-苯基-3-(2-环己酮基)-丙酮。
实施例8
用称量天平称取0.0055mol环己酮,0.0055molnaoh(固体、粒状),25ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-丁基-咔唑基)-3-(对溴苯基)-丙烯酮,搅拌并缓慢升温至75℃,使其在75℃的温度及的80rad/min搅拌速度下回流反应35min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml5℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在30℃真空干燥20h,即得到1-(3-n-丁基-咔唑基)-3-(对溴苯基)-3-(2-环己酮基)-丙酮。
实施例9
用称量天平称取0.0052mol环己酮,0.0052molnaoh(固体、粒状),22ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-戊基-咔唑基)-3-(对硝基苯基)-丙烯酮,搅拌并缓慢升温至72℃,使其在72℃的温度及的65rad/min搅拌速度下回流反应60min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml2℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在22℃真空干燥28h,即得到1-(3-n-戊基-咔唑基)-3-(对硝基苯基)-3-(2-环己酮基)-丙酮。
实施例10
用称量天平称取0.0058mol环己酮,0.0058molnaoh(固体、粒状),28ml无水乙醇加入到干燥的装有冷凝回流管和搅拌装置的三口瓶中,使其在室温下搅拌5min,混合均匀后,再加入0.001mol的1-(3-n-己基-咔唑基)-3-(对羟基苯基)-丙烯酮,搅拌并缓慢升温至78℃,使其在78℃的温度及的75rad/min搅拌速度下回流反应50min。反应期间用薄层色谱监测反应进程,选用体积比为v(乙酸乙酯):v(石油醚)=1:3的展开剂。反应完全后,停止反应,使反应体系缓慢降到室温,加入30ml4℃冷水,静置,有固体析出,抽滤,滤饼反复水洗至中性后用无水乙醇重结晶,旋转蒸发仪蒸去溶剂,再在28℃真空干燥22h,即得到1-(3-n-己基-咔唑基)-3-(对羟基苯基)-3-(2-环己酮基)-丙酮。
除上述实施例外,原料1-(3-n-取代-咔唑基)-3-芳基-丙烯酮还可采用以下物质,其中取代基为庚基、辛基、壬基、苯氧亚甲基、对氯苯氧亚甲基、邻氯苯氧亚甲基、2,4-二氯苯氧亚甲基、间氯苯氧亚甲基、对氟苯氧亚甲基、对溴苯氧亚甲基、对碘苯氧亚甲基、对甲氧基苯氧亚甲基、2-硝基苯氧亚甲基、苯氧乙基、α-萘氧亚甲基、β-萘氧亚甲基或β-萘氧乙基;芳基为间硝基苯基、邻羟基苯基、乙烯基苯基、邻甲基苯基、间甲基苯基、间甲氧基苯基、2-氟苯基、间氨基苯基、邻氨基苯基、对氨基苯基、邻氯苯基、2,4-二氯苯基、3,5-二硝基苯基、邻溴苯基、间溴苯基或呋喃基;在此不一一列举。
另外,本发明中的1-(3-n-取代-咔唑基)-3-芳基-丙烯酮可采用以下方法制备:
1)以乙酰基咔唑为原料制备n-取代-咔唑;
2)以n-取代-咔唑为原料制备3-乙酰基-n-取代-咔唑;
3)以3-乙酰基-n-取代-咔唑和芳香醛为原料,以naoh/k2co3为催化剂进行研磨反应,反应完毕后反复水洗、干燥得到粗品,粗品经硅胶柱层析法进行分离提纯,即得到1-(3-n-取代-咔唑基)-3-芳基-丙烯酮。
本发明探索了含新的取代基团的咔唑基michael加成产物以及制备方法,并优化了实验条件,缩短了反应时间,为绿色合成做出了贡献。本发明提供的1-(3-n-取代-咔唑基)-3-芳基-3-(2-环己酮基)-丙酮,是一系列全新的酮类化合物,具有杀菌、抑菌、除草等功效,可用于医药领域和农药领域等领域。
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效结构变换,均仍属于本发明技术方案的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!