一种丙炔酸类化合物的制备方法与流程
本发明涉及co2的活化转化及相关化学技术领域,涉及到一种丙炔酸类化合物的制备方法。
背景技术:
二氧化碳是储量丰富、廉价易得且可再生的c1资源,将其活化转化生成高附加值的精细化学品的研究已经引起了人们的广泛关注。在过去的几十年中,人们报道了很多关于二氧化碳固定及转化的方法[参见:(a)sakakura,t.;choi,j.-c.;yasuda,h.chem.rev.2007,107,2365.(b)q.liu;l.wu;r.jackstell;m.beller,nat.commun.2015,6,5933.]。丙炔酸类化合物是重要的合成中间体,广泛用于合成精细化学品、医药分子等。故而丙炔酸类化合物的合成一直受到广泛的关注。现有技术中,合成丙炔酸类化合物的方法主要是炔烃的氧化羧基化反应,以甲醛或一氧化碳作为羧基化试剂,但是该方法存在co毒性大等问题。近期有文献报道利用过渡金属催化或者碳酸铯促进co2与端炔反应制备丙炔酸类化合物,但是存在过渡金属催化剂价格昂贵、配体庞大且合成困难、所用高沸点溶剂后处理难等诸多问题[参见(a)dingyiyu;yugenzhang,pnas,2010,47,20189.(b)haocheng;beizhao;yingmingyao;chengronglu.greenchem.,2015,17,1675;(c)manojtrivedi;aabhinavkumarb;nigamp.rath.daltontrans.,2015,44,20874;(d)seunghyokim;kwangheekim;soonhyeokhong.angew.chem.int.ed.2014,53,771;(e)xiao-huanliu;jian-gongma;zhengniu;guang-mingyang;pengcheng.angew.chem.int.ed.2015,54,988]。也曾有报道以二氧化碳和端炔为原料在无金属催化体系中生成丙炔酸类化合物,但是所用的碱tbd以及碳酸铯比较昂贵,且使用的dmf等溶剂后处理困难[参见文献(a)yudingyi,zhangyugen,greenchem.,2011,13,1275;(b)x.wang,y.n.lim,c.lee,h.-y.jang,b.y.lee,eur.j.org.chem.2013,1867]。因此无金属催化剂、成本低且易于后处理的丙炔酸类化合物的新方法具有很好的应用前景。
技术实现要素:
本发明提供了一种无金属催化剂、无配体的体系促进co2和端炔反应生成丙炔酸类化合物的方法。该方法具有反应成本低、实验操作简单、易实现工业化等优点。
本发明是一种以端炔和co2为原料,在添加剂和碱促进下,端炔与二氧化碳在有机溶剂中反应,生成丙炔酸类化合物,反应方程式如下:
该方法采用的技术方案如下:
丙炔酸的合成:在手套箱内将添加剂、碱、溶剂以及端炔依次加入到高压釜后,在手套箱外充入co2。封闭高压釜,放入油浴锅中开始反应。
其特征在于,有机溶剂包括:甲苯、正己烷、四氢呋喃、1,4-二氧六环、二氯甲烷、三氯甲烷、乙腈。优先四氢呋喃、乙腈、1,4-二氧六环。
反应温度范围为20℃~150℃,优选75℃~100℃。
反应时间范围为4~36小时,优选15~24小时。
碱选自碳酸钾、碳酸钠、乙酸钠、乙酸钾等。优选碳酸钾。
添加剂选自甲基醋酸铵、四丁基氯化铵、四丁基溴化铵、四丁基碘化铵、四丁基醋酸铵、四正辛基溴化铵、十六烷基三甲基溴化铵、十烷基三甲基溴化铵、四丁基硝酸铵、四甲基碘化铵、三氟乙酸钠、硫酸钠、乙酸铵、甲基三苯基溴化膦、四氟硼酸三叔丁基膦、硝酸铵、硝酸钠、硝酸钾、亚硫酸钠等。优选四丁基醋酸铵和四丁基硝酸铵。
有机溶剂的量为3~10ml。
端炔化合物与碱的摩尔比为1:0.5~10,优选1:4。
端炔化合物与添加剂的摩尔比为1:0.1~5,优选1:1.5。
co2压力为0.1mpa~6mpa,优选1mpa。
附图说明
附图说明
图1是实施例1中苯丙炔酸的1h核磁谱图。
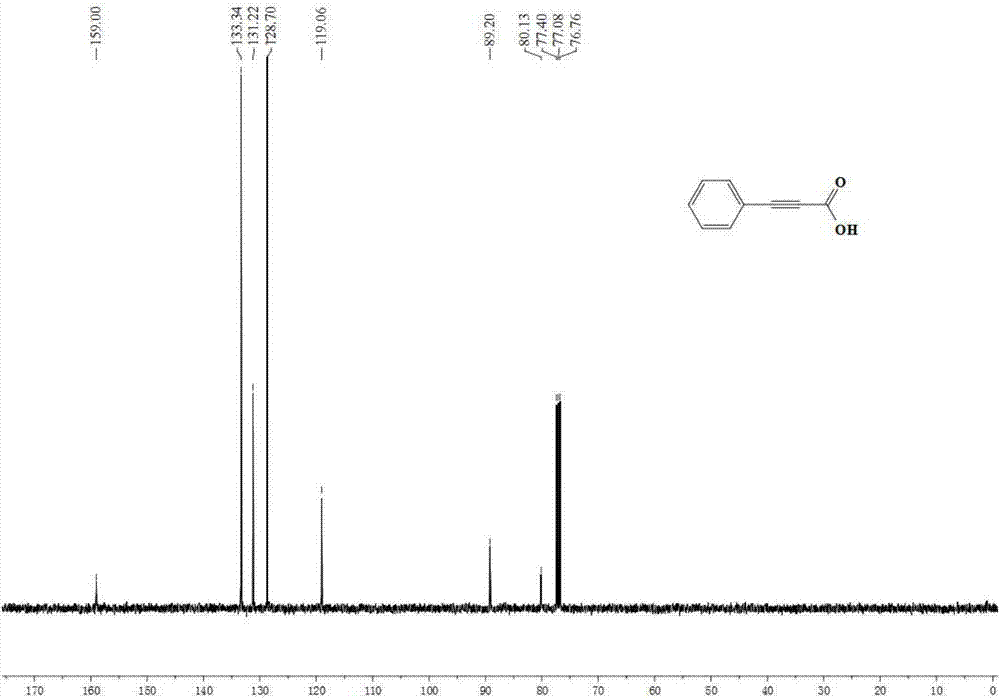
图2是实施例1中苯丙炔酸的13c核磁谱图。
图3是实施例2中2-甲氧基苯丙炔酸的1h核磁谱图。
图4是实施例2中2-甲氧基苯丙炔酸的13c核磁谱图。
图5是实施例3中4-氯本丙炔酸的1h核磁谱图。
图6是实施例3中4-氯本丙炔酸的13c核磁谱图。
图7是实施例4中3-溴苯丙炔酸的1h核磁谱图。
图8是实施例4中3-溴苯丙炔酸的1h核磁谱图。
图9是实施例5中对溴苯丙炔酸的1h核磁谱图。
图10是实施例5中对溴苯丙炔酸的13c核磁谱图。
图11是实施例6中3-氟苯丙炔酸的1h核磁谱图。
图12是实施例6中3-氟苯丙炔酸的13c核磁谱图。
图13是实施例7中4-乙基苯丙炔酸的1h核磁谱图。
图14是实施例7中4-乙基苯丙炔酸的13c核磁谱图。
图15是实施例8中2-丙炔酸噻吩的1h核磁谱图。
图16是实施例8中2-丙炔酸噻吩的13c核磁谱图。
图17是实施例9中4-丙基苯丙炔酸的1h核磁谱图。
图18是实施例9中4-丙基苯丙炔酸的13c核磁谱图。
图19是实施例10中对氟苯丙炔酸的1h核磁谱图。
图20是实施例10中对氟苯丙炔酸13c核磁谱图。
具体实施方式
本发明提供了一种在碱促进下生成丙炔酸方法,该方法具有反应绿色、实验操作简单、易实现工业化等优点,展现出良好的应用前景。
下面结合具体实施例,进一步阐述本发明。实施例仅用于说明本发明而不用于限制本发明的范围。在本领域内的技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案之内。
实施例1:苯丙炔酸的合成
在手套箱中准确量取碳酸钾(552mg,4mmol,400mol%)、四丁基醋酸铵(451.5mg,1.5mmol,150mol%)、精制过的乙腈(5.0ml)、苯乙炔(102mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(0.1mpa)。封闭反应釜,置于90℃油浴中反应4h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到苯丙炔酸124mg,收率为85%。1hnmr(400mhz,cdcl3)δ11.03(s,1h),7.65–7.57(m,2h),7.53–7.34(m,1h),7.22(d,2h).13cnmr(101mhz,cdcl3)δ159.00,133.34,131.22,128.70,119.06,89.20,80.13.
实施例2:2-甲氧基苯丙炔酸的合成
在手套箱中准确量取碳酸钠(312mg,3mmol,300mol%)、四丁基硝酸铵(304mg,1mmol,100mol%)、精制过的thf(5.0ml)、2-甲氧基苯乙炔(132mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(4mpa)。封闭反应釜,置于30℃油浴中反应36h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到2-甲氧基苯丙炔酸158mg,收率为90%。1hnmr(400mhz,cdcl3)δ10.42(s,1h),7.65(d,1h),7.54(t,1h),7.04(dd,2h),4.01(s,3h).13cnmr(101mhz,cdcl3)δ161.90,158.72,135.22,132.87,120.60,110.91,108.40,86.20,84.07,55.88.
实施例3:4-氯苯丙炔酸的合成
在手套箱中准确量取碳酸钾(552mg,4mmol,400mol%)、正四丁基溴化铵(644.6mg,2mmol,200mol%)、精制过的二氯甲烷(5.0ml)、4-氯苯乙炔(136.5mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(2mpa)。封闭反应釜,置于60℃油浴中反应18h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到4-氯苯丙炔酸145mg,收率为80%。1hnmr(400mhz,meod)δ7.56(d,2h),7.44(d,2h);13cnmr(101mhz,meod)δ155.02,136.63,133.91,128.85,118.28,83.73,81.37.
实施例4:4-甲基苯丙炔酸的合成
在手套箱中准确量取乙酸钠(123mg,1.5mmol,150mol%)、四丁基醋酸铵(451.5mg,1.5mmol,150mol%)、精制过的正己烷(5.0ml)、对甲基苯乙炔(116mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(0.5mpa)。封闭反应釜,置于100℃油浴中反应24h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到对甲基苯丙炔酸147mg,收率为92%。1hnmr(400mhz,cdcl3)δ7.44(d,2h),7.30(d,2h),2.37(s,3h).13cnmr(101mhz,cdcl3)δ159.10,142.01,133.36,129.49,115.97,89.86,79.84,21.78.
实施例5:对溴苯丙炔酸的合成
在手套箱中准确量取碳酸钾(552mg,4mmol,400mol%)、四正辛基溴化铵(2734.0mg,5mmol,500mol%)、精制过的乙腈(5.0ml)、对溴苯乙炔(181mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(1.5mpa)。封闭反应釜,置于110℃油浴中反应28h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到对溴苯丙炔酸178mg,收率为79%。1hnmr(400mhz,meod)δ7.57(d,2h),7.46(d,2h);13cnmr(101mhz,meod)δ154.98,134.00,131.86,124.91,118.69,83.78,81.49.
实施例6:3-氟苯丙炔酸的合成
在手套箱中准确量取碳酸钾(345.5mg,2.5mmol,250mol%)、四甲基醋酸铵(226.4mg,2mmol,200mol%)、精制过的1,4-二氧六环(5.0ml)、3-氟苯乙炔(120mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(3mpa)。封闭反应釜,置于100℃油浴中反应18h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到3-氟苯丙炔酸137mg,收率为84%。1hnmr(400mhz,meod)δ7.49–7.38(m,2h),7.33(d,j=9.2hz,1h),7.25(t,j=8.4hz,1h).13cnmr(101mhz,meod)δ161.11,154.89,130.57,128.56,121.47,119.00,117.79,83.30,81.10.
实施例7:4-乙基苯丙炔酸的合成
在手套箱中准确量取碳酸钾(221mg,1.6mmol,160mol%)、四丁基醋酸铵(451.5mg,1.5mmol,150mol%)、精制过的thf(5.0ml)、4-乙基苯乙炔(130mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(2mpa)。封闭反应釜,置于80℃油浴中反应20h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到4-乙基苯丙炔酸149mg,收率为86%。1hnmr(400mhz,cdcl3)δ9.62(s,1h),7.55(d,2h),7.25(t,2h),2.70(q,2h),1.26(t,3h).13cnmr(101mhz,cdcl3)δ158.79,148.15,133.47,128.30,116.18,89.75,79.80,29.05,15.14.
实施例8:2-丙炔酸噻吩的合成
在手套箱中准确量取碳酸钾(552mg,4mmol,400mol%)、甲基三苯基溴化膦(1074.7mg,3mmol,300mol%)、精制过的乙腈(5.0ml)、2-乙炔基噻吩(108mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(1mpa)。封闭反应釜,置于120℃油浴中反应12h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到2-丙炔酸噻吩118.6mg,收率为78%。1hnmr(400mhz,meod)δ7.69(d1h),7.60–7.55(m,1h),7.15(t,1h);13cnmr(101mhz,meod)δ155.08,136.21,131.26,127.48,119.04,84.63,79.00.。
实施例9:4-丙基苯丙炔酸的合成
在手套箱中准确量取碳酸钾(552mg,4mmol,400mol%)、四丁基醋酸铵(451.5mg,1.5mmol,150mol%)、精制过的乙腈(5.0ml)、4-丙基苯乙炔(144mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(1.3mpa)。封闭反应釜,置于50℃油浴中反应24h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到4-丙基苯丙炔酸165mg,收率为88%。1hnmr(400mhz,cdcl3)δ10.81(s,1h),7.56(d,2h),7.23(d,2h),2.64(t,2h),1.67(dd,2h),0.96(t,3h).13cnmr(101mhz,cdcl3)δ158.91,146.65,133.37,128.87,116.22,89.77,79.86,38.13,24.16,13.72.
实施例10:对氟苯丙炔酸的合成
在手套箱中准确量取碳酸钾(552mg,4mmol,400mol%)、四丁基醋酸铵(451.5mg,1.5mmol,150mol%)、精制过的甲苯(5.0ml)、对氟苯乙炔(120mg,1mmol),依次加入到25ml的反应釜中,手套箱外充入co2(0.7mpa)。封闭反应釜,置于75℃油浴中反应16h。反应结束后,将反应釜缓慢冷却至室温,然后缓慢放出剩余的气体。反应釜中剩余的反应液转移至单口瓶,加入1m盐酸酸化至ph=1,用乙酸乙酯萃取,收集有机相,真空除去溶剂,经硅胶柱分离(洗脱剂:石油醚/乙酸乙酯=6/1)得到对氟苯丙炔酸126mg,收率为77%。1hnmr(400mhz,meod)δ7.71(dd,2h),7.32(t,2h);13cnmr(101mhz,meod)δ165.18,162.68,155.16,135.05,134.96,115.93,115.70,84.09,80.43。
- 还没有人留言评论。精彩留言会获得点赞!