一种蝎毒素多肽及其合成方法与流程
本发明涉及蛋白质多肽药物领域,具体地说涉及一种蝎毒素多肽及其合成方法。
背景技术:
天然蝎毒素是一种发现于蝎子毒液,并经分离纯化得到的毒素蛋白,该蛋白具有治疗自身免疫性疾病的潜在药物可能,因此该毒素蛋白同时具有药物开发的潜能。
目前获得蝎毒素可采用的方法主要有:
一种是直接从蝎子来源的粗毒中提取,经过反相高效液相色谱c18柱分离纯化,从中分离出毒液中多种单一的组分。这种方法可以获得天然蝎毒素多肽,但是缺点在于得到的蝎毒素量非常微小,1毫克粗毒经过分离纯化,仅仅可以获得微克级目的毒素多肽,同时对于大规模生产操作不便。
还有一种是采用化学合成来制备,这种方法在自动化肽合成仪上进行固相肽合成获取粗制直链产物,需要再经过二硫键重构和hplc纯化过程,才能得到有活性的蝎毒素多肽。但是对于含有多对二硫键的蛋白在复性的过程中往往容易发生错配,致使复性效率较低。
技术实现要素:
本发明所要解决的技术问题是提供一种稳定性好,活性高的蝎毒素多肽。
为了解决上述技术问题,本发明采用如下技术方案:一种蝎毒素多肽,所述蝎毒素多肽的氨基酸序列如seqidno.1所示,且其中第8位与第28位之间的s-s键被替换为c-s键。
本发明还提供上述蝎毒素多肽的合成方法,包括以下步骤:
采用fmoc固相合成法,以树脂作为固相载体,经缩合剂活化,按照seqidno.1所示的序列将氨基酸由c端向n端逐个偶联到树脂上,且其中第28位cys由式1所示的二氨基二酸构件单元所替换,第28位二氨基二酸构件单元耦合完后,由c端向n端继续耦合直至第9位,待第9位耦合完后,先脱除alloc保护基团和fmoc保护基团,再令第28位二氨基二酸构件单元与第9位氨基酸进行缩合成环,最后继续耦合第7位以及余下的氨基酸;
最后经切割液处理以及复性后,获得所述蝎毒素多肽;
进一步地,所述树脂为rinkamamideresin或者2-cl-trt-cl-resin。这两种树脂来源广,成本低,而且在实施本发明的过程中,发明人发现采用这两种树脂,产率更高。
进一步地,所述缩合剂选自dic、dcc、hatu、hbtu、pyaop、pybop中的任意一种或任意几种。这里的几种是指两种以上。
进一步地,合成温度为25~90℃。在实施本发明的过程中,发明人发现在这个温度区间内,可以试用于所有氨基酸的反应条件。
进一步地,alloc保护基团的脱除试剂为四(三苯基膦)钯与苯硫酚的混合物。
进一步地,fmoc保护基团的脱除试剂为含有20vt%哌啶的dmf溶液。
进一步地,所述切割液为三氟乙酸、苯酚、水、tips、茴香硫醚、乙二硫醇的混合液。
本发明有益效果为:
本发明蝎毒素多肽具有非常稳定的结构,通过在肽片段的合成中引入c-s键替换容易断裂s-s键,不仅可以起到稳固蛋白空间构象的作用,也可以促进复性过程的剩余二硫键的配对提高整体复性效率。另外本发明方法操作简便,收率高,获得的蝎毒素多肽具有更好的稳定性,以及较高的活性,几乎和天然蝎毒素相同。
附图说明
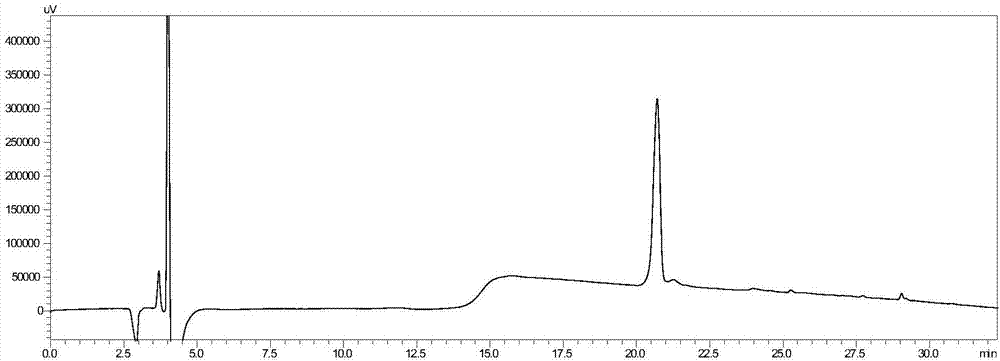
图1是本发明蝎毒素多肽复性前hplc色谱图。
图2是本发明蝎毒素多肽复性后hplc色谱图。
图3是本发明蝎毒素多肽复性前质谱图。
图4是本发明蝎毒素多肽复性后质谱图。
图5是本发明蝎毒素多肽对kv1.x家族的电流选择性的结果图(图中bar表示0.5na,40ms)。
图6a和图6b是本发明蝎毒素多肽对kv1.3钾离子通道抑制效果及ic50曲线图。
具体实施方式
下面将参考附图并结合实施例来详细说明本发明。
实施例1
二氨基二酸构件单元的制备
反应式为:
具体步骤为:
化合物2:取1.5g(12.6mmol)高丝氨酸和1.34g(12.6mmol)碳酸钠于150ml圆底烧瓶中,用30ml水和15ml1,4-二氧六环溶解。反应体系在0℃下搅拌,分批加入4.26g(12.6mmol)芴甲氧羰酰琥珀酰亚胺(fmoc-osu)。添加完毕后,使反应体系恢复至室温(若反应体系有固体未溶解,可适量多加入1,4-二氧六环使其溶解),搅拌过夜。用旋转蒸发仪除去溶剂中的1,4-二氧六环,之后用乙酸乙酯萃取剩余水相。保留水相,用6m的盐酸溶液调节ph至2。用乙酸乙酯萃取调节ph后的水相,合并有机相并用饱和食盐水洗,无水硫酸钠干燥。过滤有机相,用旋转蒸发仪除去溶剂,得到白色固体。将白色固体转移至250ml带抽气支管的反应瓶中,氮气保护下加入用75ml二氯甲烷和18ml无水四氢呋喃使固体溶解。反应体系中加入叔丁基三氯乙酰亚胺酯4.5ml(5.51g,25.2mmol)。反应体系在氮气保护下,室温搅拌过夜。旋转蒸发仪除去溶剂,得到的固体用乙酸乙酯溶解后,用饱和碳酸氢钠水溶液洗涤有机相,再用饱和食盐水洗涤,无水硫酸钠干燥。将干燥后的有机相过滤,粗硅胶(100-200目)拌样,旋转蒸发仪旋干溶剂。柱层析分离法(硅胶柱)分离目的产物,淋洗剂乙酸乙酯:石油醚=1:3。得化合物2为无色油状物,2.6g,产率52%。
化合物3:取2.86g(7.2mmol)化合物2和2.85g(8.6mmol)四溴化碳,用26ml二氯甲烷溶解于150ml的圆底烧瓶中。将反应瓶置于冰浴中。取2.26g(8.6mmol)三苯基膦溶解于13ml二氯甲烷中,0℃下用恒压滴液漏斗逐滴加入反应体系中。滴加完毕后,使反应体系恢复至室温,反应2小时。反应完毕后,加入100ml二氯甲烷稀释。分别用水和饱和食盐水洗涤,无水硫酸钠干燥。将有机相过滤,用旋转蒸发仪除溶剂。用15ml二氯甲烷溶解得到的残余物,转移至500ml烧杯中,剧烈搅拌下用乙酸乙酯:石油醚=1:1溶剂400ml溶解,搅拌十分钟。此过程中,会有大量白色固体三苯氧磷析出。将固体滤出,得到的有机相用旋转蒸发仪除去溶剂。用适量乙酸乙酯溶解得到的残余物,粗硅胶(100-200目)拌样,旋转蒸发仪旋干溶剂,柱层析分离法(硅胶柱)分离目的产物,淋洗剂乙酸乙酯:石油醚=1:6。得化合物3为微黄色油状物,2.81g,产率85%。
化合物4:称取2.0g(5.51mmol)trt-半胱氨酸和0.58g(5.51mmol)碳酸钠置于100ml茄型瓶中,用10ml水和5ml乙腈溶解并将反应瓶放置于冰浴中。取0.58ml(5.51mmol)氯甲酸丙烯酯,0℃下逐滴加入至反应体系。滴加完毕后,使反应恢复室温,搅拌过夜。反应体系中的乙腈用旋转蒸发仪除去,剩余溶液加入20ml水,用6m盐酸溶液调节ph至2(调节过程中会有固体产生,可边加盐酸边加入乙酸乙酯搅拌,使最终水相的ph为2即可)。用乙酸乙酯萃取水相中的有机物,合并有机相后用旋转蒸发仪除去乙酸乙酯。得到的剩余物用15mldmf溶解,加入碳酸氢钠0.46g(5.51mmol)。取烯丙基溴0.58ml(6.61mmol)加入至反应体系中。反应体系在室温下搅拌约20小时。反应完毕后,用旋转蒸发仪除去溶剂,剩余物用乙酸乙酯溶解后,分别用水、饱和碳酸氢钠溶液、水、0.1m硫酸氢钾溶液和饱和食盐水洗涤,无水硫酸钠干燥。过滤,用粗硅胶(100-200目)拌样,旋转蒸发仪旋干溶剂,柱层析分离法(硅胶柱)分离目的产物,淋洗剂乙酸乙酯:石油醚=1:5。得化合物4为无色油状物,2.0g,产率74%。
化合物5和化合物6:取1.68g(3.45mmol)化合物4用35ml二氯甲烷溶于250ml圆底烧瓶中,加入6ml三氟乙酸和1.4ml三异丙基硅烷。反应在室温下搅拌2小时。将反应倒入500ml锥形瓶中,加入100ml二氯甲烷和50m水。搅拌下,缓慢加入碳酸氢钠固体中和过量的酸。分离得有机相,用饱和食盐水洗涤,无水硫酸钠干燥。过滤有机相,旋转蒸发仪除去溶剂,得到化合物5。迅速地将化合物5用10ml乙酸乙酯溶解,倒入250ml带抽气支管的圆底烧瓶中。取1.91g(4.15mmol)化合物3用10ml乙酸乙酯溶解,氮气保护下,加入化合物5的乙酸乙酯溶剂中(该步反应溶剂乙酸乙酯在使用前,用超声振荡器去除里面的气体,再用氮气鼓泡,确保除掉乙酸乙酯中的氧气)。加入4.47g四丁基溴化铵和16ml饱和碳酸氢钠溶液。氮气保护下,室温搅拌20小时。加入50ml水,并分离水相和有机相。水相用乙酸乙酯萃取,合并有机相。分别用水和饱和食盐水洗涤有机相,无水硫酸钠干燥。过滤,用粗硅胶(100-200目)拌样,旋转蒸发仪旋干溶剂,用柱层析分离法(硅胶柱)分离目的产物,淋洗剂乙酸乙酯:石油醚=1:5。得化合物6为无色油状物,1.94g,产率90%。
化合物1:取1.14g(1.82mmol)化合物用6ml二氯甲烷溶解置于50ml圆底瓶中,加入6ml三氟乙酸和258μl三异丙基硅烷。室温下搅拌2小时。反应液转移至250ml的茄形瓶中,加入100ml二氯甲烷。旋转蒸发仪去除溶剂,剩余少量液体时,重复加入二氯甲烷并旋蒸去除溶剂的操作数次以除去大部分残余的三氟乙酸。用乙腈:水=1:1溶解得到的剩余物,装入ep管中,用液氮将其充分冻结后放入冻干机冻干。得到化合物1为白色粉末状固体,1.90g,产率98%,也即为二氨基二酸构件单元。
化合物2核磁共振-氢谱(cdcl3,400mhz):7.77(d,2h);7.60(d,2h);7.41(t,2h);7.32(t,2h);5.62(d,1h);4.44(m,2h);4.22(t,1h);3.69(dd,1h);3.59(dd,1h);1.48(s,9h)。高分辨质谱:398.19598ftms-esi[m+h]+(calculatedforc23h28no5)
化合物3高分辨质谱:482.09399ftms-esi[m+na]+(calculatedforc23h26brno4na)
化合物4核磁共振-氢谱(cdcl3,400mhz):7.39(d,6h);7.28(m,6h);7.21(m,3h);5.88(m,2h);5.26(m,5h);4.60(m,1h);4.54(d,1h);2.67(dd,1h);2.57(dd,1h)。
化合物6核磁共振-氢谱(cdcl3,400mhz):7.76(d,2h);7.60(d,2h);7.40(t,2h);7.32(t,2h);5.89(m,2h);5.44-5.18(m,4h);4.65(d,2h);4.58(m,2h);4.39(m,3h);4.23(t,1h);3.01(d,2h);2.57(m,2h);2.09(m,1h);1.91(m,1h);1.49(s,9h)。
化合物1核磁共振-氢谱(dmso-d6,400mhz):7.90(d,2h);7.72(d,2h);7.42(t,2h);7.33(t,2h);5.89(m,2h);5.30(m,2h);5.19(t,2h);4.59(d,2h);4.49(d,2h);4.28(d,2h);4.23(m,2h);4.08(m,1h);2.92(dd,1h);2.77(dd,1h);1.91(m,2h)。质谱ms569.20[m+h]+(calculatedforc29h33n2o8s);591.18[m+na]+(calculatedforc29h32n2o8sna)。
实施例2
蝎毒素多肽的制备
所述蝎毒素多肽的氨基酸序列如seqidno.1所示,且其中第8位与第28位之间的s-s键被替换为c-s键,也即相当于其中第28位cys上的s原子由c原子所替换。
反应式为:
具体步骤为:
称取rinkamamideresin(0.1mmol)于固相合成管中,使用dmf/dcm混合溶剂(体积比为1:1)溶胀15min后,抽干,然后按照高温辅助的fmoc固相合成法合成上述蝎毒素多肽(0.4mmol氨基酸,0.4mmoloxyma,0.4mmoldic;反应时间为20-30min,反应温度为75℃),其中精氨酸(arg)采用hctu作为缩合剂,室温反应40min2次;组氨酸(his)采用dic作为缩合剂,室温反应40min;半胱氨酸(cys)采用dic作为缩合剂,50℃反应40min;
其中第28位cys由化合物1,也即二氨基二酸构件单元所替换,第28位二氨基二酸构件单元耦合完后,由c端向n端继续耦合直至第9位,待第9位耦合完后,先采用四(三苯基膦)钯与苯硅烷的混合物(摩尔比1:5)作为脱除试剂脱除其alloc保护基团,采用含有20vt%哌啶的dmf溶液作为脱除试剂脱除其fmoc保护基,再令第28位二氨基二酸构件单元与第9位氨基酸进行缩合成环,最后继续耦合第7位以及余下的氨基酸;
待所有的氨基酸耦合完后,用dmf,dcm,dmf各洗5次,加入含有哌啶(体积分数20%)的dmf溶液反应2次(2min,10min各一次),脱除fmoc保护基;
组装完肽链后,加入8ml切割试剂[v(tfa):v(苯酚):v(水):v(三异丙基硅烷,tips)=88:5:5:2]于多肽合成管内,反应2h,将多肽从树脂上切下,并脱除侧链保护基团,将切割液聚集于离心管中,用n2鼓泡,浓缩溶液,最后用冰乙醚沉淀,离心,得到粗肽,使用半制备型hplc分离得到纯肽,进一步经过复性,具体步骤为将1mg纯肽溶于含有2mm还原型谷光氨肽,0.2mm氧化型谷胱甘肽的pbs(6m盐酸胍,0.2mm磷酸氢二钠,ph8.0)缓冲液中,至终浓度为0.25mg/ml,4摄氏度复性过夜后,经hplc分离,得到所述蝎毒素多肽。
该蝎毒素多肽复性前后的hplc色谱图和质谱图参见图1至图4,从图中可以看出,复性之后的蝎毒素多肽的极性要略大于未复性之前的。
实施例3
蝎毒素多肽的活性测试
cho细胞的培养以及转染过程
中国仓鼠卵巢(cho)细胞,没有钾离子通道的本底电流,在研究钾离子通道时不会造成干扰。
细胞培养基:培养细胞使用的dmem/f12培养基(gibco公司)。使用时前加入10%的胎牛血清(fbs)(gibco公司)以及100u/ml的青霉素和链霉素(gibco公司)。
细胞复苏:打开37摄氏度的水浴锅。配制含有血清fbs和双抗青霉素与链霉素的dmem/f12培养基,并置于37摄氏度水浴锅,预热至37摄氏度。将细胞从液氮灌中取出,迅速放入37摄氏度的水浴锅,待其融化后,放入离心机中,1000rpm离心5min,弃去上清。用1ml预热好的dmem/f12培养基将细胞重悬,加入60mm培养皿中,补加2ml培养基,混合均匀后放入37摄氏度,5%co2培养箱中培养。24h后在显微镜下观察细胞状态,此时贴壁细胞为存活的细胞,死亡的细胞则漂浮在培养基中。
细胞传代:待细胞汇合度大约在90%左右,即可进行细胞传代。在超净台中将60mm细胞培养皿中的上清完全吸走,加入1mlpbs清洗贴壁的细胞一次,吸走pbs。然后加入1ml0.125%的胰酶(含0.01%的edta)消化1min30s吸走胰酶。加入3mldmem/f12培养基终止消化,轻轻吹打重悬培养皿底部的细胞,按照5:1的比例进行传代,吸取0.6ml细胞悬液到新的60ml培养皿中,补加3ml新鲜的培养基,混匀后放入37摄氏度,5%co2培养箱中进行培养,待长满后再次进行传代。
细胞铺空以及转染:细胞传代时多余的细胞悬液取200μl加入24孔板的一个小孔中,补加300μldmem/f12培养基,在37摄氏度,5%co2培养箱中培养24h,即可用于质粒转染。
采用脂质体转染的方式。转染时取出24孔板,将原来的0.5ml培养基吸走,用0.2ml的pbs清洗一遍,洗去残留的血清和抗生素,然后加入0.4mlopti-mem培养基(无抗生素,无血清)。取两个无菌的ep管,分别加入2μl阳离子脂质体lipofectamine2000(invitrogen)以及0.6μg的pcdna3.1-egfp质粒,1.0μg的pcdna3.1-kv1.3质粒,然后用50μl的opti-mem培养基稀释。稀释好后在超净台中室温静置5min,使质粒和脂质体在培养基中混合均匀。然后将质粒和脂质体混合到一起,静置20min,使阳离子脂质体将质粒包裹起来形成转染复合物。然后将这100μl的混合物滴加到24孔板中,放入37摄氏度5%co2培养箱中培养4-6h。
转染完成后,需要将细胞转移到玻璃片上进行后续的膜片钳电生理实验。玻璃片大小约为0.8cm×0.8cm,用洗洁精、稀盐酸、ddh2o清洗后,浸泡在酒精中。使用时用镊子夹出一片,在酒精灯上灼烧之后,放入35mm的小皿中。然后在玻片上滴加poly-l-lysine(用ddh2o配制成0.05mg/ml的溶液),由于poly-l-lysine对细胞有一定的毒性,因此过3min吸去它之后可以再在玻片上滴加ddh2o进行清洗,晾干备用。然后将24孔板从培养箱中取出,吸去含有脂质体和质粒的opti-mem培养基,加入200μl,0.125%的胰酶(含0.01%edta)消化20s,迅速吸走胰酶,加入600μldmem/f12培养基终止消化,吹打孔底部,重悬细胞。重悬后将细胞悬液滴加到玻璃片上。将35mm的培养皿放入培养箱中,1h后等到玻片上的细胞贴壁后,补加2mldmem/f12培养基。继续放入培养箱中培养,24h-48h之间即可用于膜片钳电生理实验。
电生理实验
电极拉制:我们使用的电极拉制仪是美国sutter公司的p-1000水平拉制仪,玻璃电极是sutter公司的硼硅酸盐玻璃毛细管,外径1.50mm,内径0.86mm。使用电极拉制仪上预设程序,使电极分四步拉制,电极的电阻维持在3-5mω。
电生理溶液:cho细胞电生理的细胞外液,它包含150mmnacl,4mmkcl,2mmcacl2,1mmmgcl2,10mmhepes。称取粉末溶于一定体积的ddh2o后,用ph计调节ph至7.4,用容量瓶定容,测定渗透压,一般为300mosm-310mosm。用于局部给药的蝎毒素多肽也用这种细胞外液配制。而电极内液包含140mmkcl,10mmnacl,5mmegta,10mmhepes同样调节ph至7.4后定容,测定渗透压,一般为290mosm-300mosm左右。细胞外液和电极内液在使用前都需要用0.22μm的滤膜过滤。
kv1.3电流刺激protocol:我们记录在cho细胞中表达的人源kv1.3野生型全细胞电流时,将电压hold在-80mv,然后给予去极化电压刺激,从-80mv~+60mv,每次增加20mv,
维持200ms,引出kv1.3钾离子电流,再回到-80mv,每个刺激之间间隔30s,以避免kv1.3发生积累失活效应。给药时,则每隔30s的时间间隔,重复从-80mv到+40mv的去极化电压刺激,观察kv1.3钾离子电流大小的变化。
电生理记录操作过程:天转染滴片后的玻璃片从培养基中取出,用细胞外液清洗一遍,然后放入一个装有2ml细胞外液的35mm培养皿底部,放到显微镜载物台的中央。用左手手动微操控制给药管,使它的尖端靠近细胞。电极尖端灌入电极内液后,固定在电极夹持器上。电极入水前,给予一个正压,主要为了防止外液中杂质进入电极内液,影响封接。然后通过右手微操下降电极,进入液面以下,调节液接电位。在显微镜中找到电极,然后将其靠近细胞,电极接触细胞时可以观察到电阻增加,此时释放正压,轻轻给予负压,即可形成gω封接,然后补偿快电容。将细胞吸破,补偿慢电容和串联电阻。待细胞稳定一段时间后,即可进行电生理记录。所使用的放大器为heka公司的epc10放大器,采集软件为patchmaster,电流记录频率为10khz,所有实验都在室温进行。
测试蝎毒素多肽活性,记录kv1.x家族电流,当电流稳定时,细胞外给蝎毒素多肽,观察蝎毒素多肽对kv1.x家族电流的作用,结果如图5所示。
使用不同浓度的蝎毒素多肽,观察不同浓蝎毒素多肽对kv1.3影响,收取蝎毒素多肽对kv1.3药效曲线,对kv1.3钾离子通道抑制效果及ic50曲线见图6a和图6b。
结果显示,蝎毒素多肽对kv1.3的有专一性选择性抑制效果,并且抑制效果浓度曲线结果表明ic50值为3nm。
综上所述,本发明获得的蝎毒素多肽具有更稳定的结构,以及和天然蝎毒素相同的活性。
应当理解本文所述的例子和实施方式仅为了说明,并不用于限制本发明,本领域技术人员可根据它做出各种修改或变化,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
sequencelisting
<110>中国科学技术大学先进技术研究院
<120>一种蝎毒素多肽及其合成方法
<130>1
<160>2
<170>patentinversion3.1
<210>1
<211>38
<212>prt
<213>odonthobuthusdoriae
<400>1
glyvalprothraspvallyscysargglyserproglncysilegln
151015
procyslysaspalaglymetargpheglylyscysmetasnglylys
202530
cyshiscysthrprolys
35
- 还没有人留言评论。精彩留言会获得点赞!