一种二甘油单缩醛及其制备方法和应用与流程
本发明涉及一种二甘油单缩醛及其制备方法和应用,属于精细化工
技术领域:
。
背景技术:
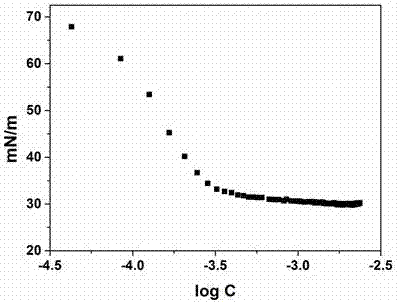
:生物柴油是现有能源框架下的有效补充性能源。随着生物柴油的广泛应用,副产物甘油的利用成为了急需解决的问题。因为生产过程中加入的甲醇使得这一部分的甘油利用受到限制,所以通过氧化、氢化、醚化、酯化、缩合等方法转化为高附加值的化学品。二甘油是两分子甘油通过自身醚化得到的含有四个羟基的多元醇醚,是甘油的低聚产物之一。由于多羟基的存在,使得其在化妆品、食品乳化剂以及聚合物增塑剂等方面均有应用。二甘油与醛或酮也可以发生缩醛化或缩酮化反应合成新型的、具有潜在应用价值的物质。二甘油具有四个羟基,醛或酮可以选择性的与两个羟基进行反应生成环状缩醛,而保留另外两个羟基,生成二甘油单缩醛的产物。专利wo9412489a1中描述了酸性催化剂对甲苯磺酸催化二甘油和羰基化合物(醛或酮)在dmf溶剂下发生缩醛化或缩酮化反应合成产物二甘油单缩醛或二甘油单缩酮,并将产物应用到洗涤剂以及洗涤剂复配上的性质。上述反应的二甘油单缩醛或二甘油单缩酮的选择性低,而且加入有机酸作为催化剂和有机溶剂,反应不易操作,对环境不友好。二甘油与醛通过缩醛化反应极易形成二甘油二缩醛,二甘油二缩醛是四个羟基全部缩醛化的产物,减少了分子的亲水性质,从而限制了其在表面活性剂上的应用。技术实现要素:本发明旨在提供一种二甘油单缩醛及其制备方法,二甘油与碳原子数为4~16的饱和脂肪族醛通过缩醛化反应合成的二甘油单缩醛,作为表面活性剂具潜在的应用价值。本发明提供了一种二甘油单缩醛,为下述(i)、(ii)、(iii)、(iv)所示四种同分异构体的混合物:其中r为c3~c15直链烷基。上述二甘油单缩醛,其中所述r优选为c5~c11的直链烷基。本发明提供了一种权利要求1或2任一项所述的二甘油单缩醛的制备方法,采用路易斯酸为催化剂,不使用任何溶剂,催化二甘油与碳原子数为4~16的饱和脂肪族醛发生缩醛化反应制备二甘油单缩醛;所述饱和脂肪族醛与二甘油的摩尔比为1:(0.2~5),优选的摩尔比是1:(0.2~2)。上述制备方法包括以下步骤:(1)将饱和脂肪族醛与二甘油混合,以醛为基准物,加入路易斯酸作为催化剂,搅拌升温至50~220℃,反应1~12小时;(2)反应结束后,经水洗、萃取、洗涤除去催化剂、干燥、真空浓缩、柱层析得二甘油单缩醛产物。上述制备方法中,所述醛与路易斯酸催化剂的质量比为1:(0.002~1)。上述制备方法中,所述路易斯酸催化剂为氯化锌、氯化钡、氯化镁、氯化钙、氯化铁、氯化铝、氯化铜或氯化亚铁中的一种。上述制备方法中,所述步骤(2)中,反应结束后,采用蒸馏水洗涤,乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂,使用无水硫酸钠干燥,使用石油醚和乙醇体积比(10-30):1柱层析得到二甘油单缩醛。上述制备方法中,所述缩醛化反应方程式为:其中r为c3~c15直链烷基。反应原理:饱和脂肪族醛和二甘油反应,在路易斯酸的作用下,金属离子能够选择性地与羟基发生作用,从而生成反应中间体,占据一部分的羟基位置,暴露另一部分的羟基,从而醛可以选择性地与二甘油发生反应,生成二甘油单缩醛。本发明提供了上述二甘油单缩醛在表面活性剂中的应用。本发明的有益效果:本发明是在无溶剂体系下由路易斯酸催化剂催化二甘油与碳原子数为4~16的饱和脂肪族醛通过缩醛化反应制备二甘油单缩醛,产品降低水溶液表面张力的能力强,泡沫丰富,乳化效果好,耐碱、耐硬水性好。附图说明图1为实施例1制备得到的二甘油单缩醛的ft-ir红外光谱。图2为实施例3制备得到的二甘油单缩醛的水溶液表面张力曲线。具体实施方式下面通过实施例来进一步说明本发明,但不局限于以下实施例。实施例1:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正辛醛:二甘油=1:1.3(摩尔比,下同)加入正辛醛4.92g,按照正辛醛:氯化锌=1:0.04(质量比,下同)加入氯化锌催化剂0.20g,在氮气氛围下,搅拌升温至100℃,反应4小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(体积比为20:1,下同)柱层析后得到二甘油缩单辛醛,收率81.2%。对产物进行红外光谱表征,产物的红外光谱如图1所示。在3441cm-1是自由oh的特征吸收峰,2924cm-1、22853cm-1、1456cm-1、1418cm-1是ch3、ch2的伸缩振动峰,11123cm-1和1045cm-1为c-o-c结构的特征峰,724cm-1是长链亚甲基中ch2的伸缩振动峰,812cm-1是六元环缩醛的特征峰,951cm-1是五元环缩醛的特征峰。质谱测得产物分子量为276。实施例2:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正丁醛:二甘油=1:0.5加入正丁醛7.20g,按照正丁醛:氯化亚铁=1:0.5加入氯化亚铁催化剂3.60g,在氮气氛围下,搅拌升温至50℃,反应12小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(10:1)柱层析后得到二甘油缩单丁醛,收率85.3%。质谱测得产物分子量为220。实施例3:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正己醛:二甘油=1:1.5加入正己醛3.33g,按照正己醛:氯化镁=1:0.07(质量比,下同)加入氯化镁催化剂0.23g,在氮气氛围下,搅拌升温至80℃,反应2小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(17:1)柱层析后得到二甘油缩单己醛,收率86.6%。质谱测得产物分子量为248。实施例4:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正十二醛:二甘油=1:5加入正十二醛1.84g,按照正十二醛:氯化钡=1:0.1加入氯化钡催化剂0.18g,在氮气氛围下,搅拌升温至150℃,反应6小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(25:1)柱层析后得到二甘油缩单十二醛,收率79.3%。质谱测得产物分子量为332。实施例5:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正癸醛:二甘油=1:2加入正癸醛3.90g,按照正癸醛:氯化钙=1:0.3加入氯化钙催化剂1.17g,在氮气氛围下,搅拌升温至120℃,反应4小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(19:1)柱层析后得到二甘油缩单癸醛,收率80.7%。质谱测得产物分子量为304。实施例6:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正戊醛:二甘油=1:0.2加入正戊醛21.50g,按照正戊醛:氯化铁=1:0.002加入氯化铁催化剂0.043g,在氮气氛围下,搅拌升温至70℃,反应9小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(15:1)柱层析后得到二甘油缩单戊醛,收率77.5%。质谱测得产物分子量为234。实施例7:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正十四醛:二甘油=1:3加入正十四醛3.53g,按照正十四醛:氯化铝=1:0.006加入氯化铝催化剂0.021g,在氮气氛围下,搅拌升温至200℃,反应3小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(27:1)柱层析后得到二甘油缩单十四醛,收率76.4%。质谱测得产物分子量为360。实施例8:在装有电磁加热器、冷凝管、氮气导入管的三口圆底烧瓶中加入二甘油8.30g,然后按正十六醛:二甘油=1:4.2加入十六醛2.86g,按照正十六醛:氯化铜=1:0.08加入氯化铜催化剂0.23g,在氮气氛围下,搅拌升温至220℃,反应5小时后冷却至室温,所得反应液经水洗涤、乙酸乙酯萃取、饱和碳酸钠溶液洗涤除去催化剂、无水硫酸钠干燥、真空浓缩,用石油醚和乙醇(30:1)柱层析后得到二甘油缩单十六醛,收率74.1%。质谱测得产物分子量为388。实施例9:性能检测1、表面张力性质:表面活性剂溶液的表面张力和临界胶束浓度(cmc)采用带有wilhelmy铂板的krussk100表面张力仪测量。参考gb/t22237-2008,在测量中,利用dosimattm剂量计向初含有60ml去离子水的容器中增加1ml0.6g/l的表面活性剂的溶液,然后充分搅拌,抽取1ml的溶液以保证测量容器中液体体积不变,测量所生成溶液的表面张力,6s读取一个数据,测量一分钟,最终取平均值。在每个浓度点重复以上的方法。实施例3的样品测试结构如图2所示。实施例3的样品在水溶液中的临界胶束溶度为3×10-4mol/l,临界胶束浓度下的表面张力γcmc=30mn/m。2、发泡性质选取实施例1所得样品,与现有技术中的工业级aeo-3(对比样品)进行比较。按照国标gb/t7462-94改进罗氏泡沫法,在25℃下测试浓度为1g/l。实施例1样品的起始泡沫高度和5min泡沫高度分别是250ml和200ml。对比样aeo-3的起始泡沫高度和5min泡沫高度分别为50ml和30ml。在40℃下测试浓度为1g/l,实施例1样品的起始泡沫高度和5min泡沫高度分别是300ml和250ml。对比样aeo-3的起始泡沫高度和5min泡沫高度分别为80ml和40ml。结果表明,来自本发明实施例1的样品,在常温和40℃发泡性质优异。3、在氢氧化钠溶液下的耐碱性测试对实施例1、实施例4和实施例5所制得的样品和与现有技术中的aeo-3(工业级)的耐碱性对比。氢氧化钠耐碱性测试:分别使用2.5wt%、5wt%或7.5wt%的氢氧化钠溶液(以质量计),加入所使用的碱溶液的0.1%wt所制得的本发明实施例样品和对比样品,搅拌10min后,静置24h,观察溶液的状态。结果如表1所示。表1样品2.5%5%7.5%对比样品aeo-3均匀浑浊不均匀浑浊不均匀浑浊实施例1无变化无变化均匀浑浊实施例4无变化均匀浑浊均匀浑浊实施例5无变化均匀浑浊均匀浑浊表1中,无变化:溶液与原溶液无明显变化。均匀浑浊:溶液比原溶液浑浊,而且均匀。不均匀浑浊:溶液与原溶液相比,有明显分层或物质析出。由表1可见,实施例1的样品加入量为2.5%和5%时,溶液无变化;在加入量为7.5%时变浑浊。实施例4和5的样品加入量为2.5%时,溶液无变化;在加入量为5%和7.5%时变浑浊。相反,对比样aeo-3在加入量为2.5wt%时变浑浊,在5%和7.5%的条件下有白色不溶物析出。结果表明,来自本发明的样品与aeo-3相比,显示出显著优异的耐碱性效果。4、抗硬水测试对实施例1、实施例4和实施例5所制得的样品和与对比样aeo-3的抗硬水性对比。抗硬水测试:分别使用1200ppm、1800ppm或2400ppm的硬水(以碳酸钙计),加入所使用的硬水的0.1%wt所制得的本发明实施例样品和对比样品,搅拌10min后,静置24h,观察溶液的状态。结果见表2所示。表2样品1200ppm1800ppm2400ppm对比样aeo-3均匀浑浊均匀浑浊不均匀浑浊实施例1无变化无变化均匀浑浊实施例4无变化均匀浑浊均匀浑浊实施例5无变化均匀浑浊均匀浑浊表2中,无变化:溶液与原溶液无明显变化。均匀浑浊:溶液比原溶液浑浊,而且均匀。不均匀浑浊:有明显分层或物质析出。由表2可见,实施例1的样品加入量为1200ppm和1800ppm时,溶液无变化;在加入量为2400ppm时变浑浊。实施例4和5的样品加入量为1200ppm时,溶液无变化;在加入量为1800ppm和2400ppm时变浑浊。相反,aeo-3在加入量为1200ppm和1800ppm时变浑浊,在2400ppm下有白色不溶物析出。结果表明,来自本发明的样品与aeo-3相比,显示出显著优异的抗硬水效果。5、乳化能力测试对制备实施例1、实施例4和实施例5所制得的样品和与对比的aeo-3的在液体石蜡的乳化能力对比。乳化能力测试:加入纯净水2wt%的所制得的实施例样品或对比样样品,再分别加入纯净水的10wt%、30wt%、50wt%的液体石蜡,超声10min,静置2h,观察乳液的状态。结果如表3所示。表3样品10%30%50%对比样aeo-3白色均相白色均相乳液分层实施例1白色均相白色均相白色均相实施例4白色均相白色均相白色均相实施例5白色均相白色均相白色均相表3中,白色均相:乳液为白色均匀的相;乳液分层:乳液明显分为两相。由表3可见,当其他条件相同,各个实施例的样品和对比样在加入量为10%和30%时,能够与基础油形成白色均相;当各个实施例和对比样aeo-3的水加入量为50%,实施例的样品仍然能够与液体石蜡形成白色均相,但对比样与液体石蜡形不成白色的均相,出现了两相明显的分层。由此可见,各个实施例样品均比aeo-3的乳化性质好。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 一种含有4‑二甲基氨基肉桂醛的耐热酚醛树脂及其制备方法与流程
- 一种二甘油单缩醛及其制备方法和应用与流程
- 一种合成3,3′‑二甲基‑4,4′‑二氨基二环己基甲烷的方法与流程
- 4‑氯‑2‑氰基‑1‑二甲基氨基磺酰基‑5‑(4‑甲基苯基)咪唑的合成方法与流程
- 3‑[(二甲基氨基)甲基]‑N‑{2‑[4‑(羟基氨基甲酰基)苯氧基]乙基}‑1‑苯并呋喃‑2‑甲酰胺的新颖的盐、有关的结晶形式、用于制备其的方法及包含其的药物组合物与流程
- 5‑溴水杨醛缩‑2‑氨基‑2‑甲基‑1,3‑丙二醇席夫碱镍配合物及合成方法与制造工艺
- (-)-(1r,2r)-3-(3-二甲基氨基-1-乙基-2-甲基丙基)-苯酚盐酸盐的晶形的制作方法