一种帕瑞昔布钠晶体化合物及其制备方法与流程
本发明属于医药
技术领域:
,公开了一种帕瑞昔布钠晶体化合物及其制备方法。
背景技术:
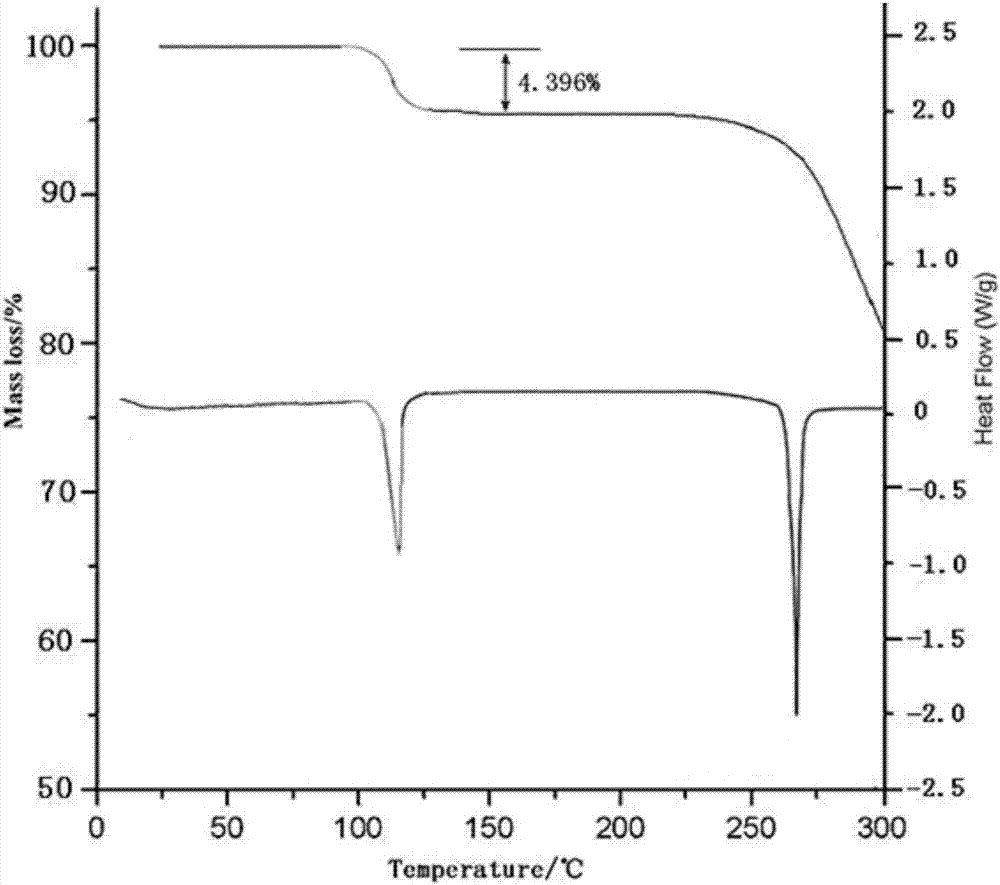
:帕瑞昔布钠(parecoxibsodium),是由辉瑞公司研制开发的第一个可静脉给药(iv)和肌肉注射(im)的特异性的cox-2抑制剂,与重磅药品塞来昔布、艾瑞昔布为同类药物,用于手术后疼痛的短期治疗,于2002年3月在欧洲上市。化学名称为:n-[[4-(5-甲基-3-苯基-4-异恶唑基)苯基]磺酰基]丙酰胺钠盐,分子式为:c19h17n2nao4s,分子量为:392.41。帕瑞昔布钠作为伐地昔布的前体药物,具有较高溶解度,容易水解为伐地昔布。尽管帕瑞昔布钠在体内迅速转化成伐地昔布而发挥作用,但由于伐地昔布的水溶性差,若在体外便转化成较多的伐地昔布,将使得溶液形成不溶的颗粒物质,影响药效的发挥还造成安全隐患。因此,在进入人体前,应确保产品全部以帕瑞昔布钠的形式存在或尽量保持伐地昔布的含量在非常低的水平。除此之外,帕瑞昔布钠的外观成形性、复溶性和含水量偏高等问题也是制剂生产中的难点。本领域技术人员都知道,药物多晶型是药物研发中的常见现象,是影响药品质量的重要因素。同一药物的不同晶型在外观、溶解度、熔点、溶出度、生物有效性等性质方面可能会有显著差异,从而直接影响药物的稳定性、生物利用度及疗效。因此,在药品研发中,应全面系统地进行晶型筛选,对晶型进行深入研究,从而找到最合适开发的晶型,以提高药物的稳定性和生物利用度,增进临床疗效。现有技术中公开了很多晶型,具体如下:专利us5932598中公开了合成帕瑞昔布钠的方法,但该专利仅公开了其熔点为271.5-272.7℃,对帕瑞昔布钠的结晶结构未进行表征。wo2003/078408a1提供了结晶形式的帕瑞考昔钠,该专利公开了三种晶型a、b、e,其中,a至少为具有至少两个选自如下2θ值的粉末x射线衍射图样:5.6、9.6、11.0和14.5±0.2度;b至少为具有至少两个选自如下2θ值的粉末x射线衍射图样:4.2、8.3、12.4、16.7、17.5、20.8和24.7±0.2度;e至少为具有至少两个选自如下2θ值的粉末x射线衍射图样:8.8、11.3、15.6、22.4、23.5和26.4±0.2度。wo2004/087682a1公开了六种晶型,其2θ分别为5.7、8.3、10.4、17.4、21.0和23.2;5.4、6.8、7.9、10.6、16.2、17.1、19.5、20.4和22.4;5.3、5.9、6.6、7.8、8.3、10.7、11.9、12.2、16.1、19.5、20.0、21.6、23.4和30.1;5.2、7.9、12.1、17.3、17.9、22.5、23.4和27.1;6.5、7.7、9.3、10.6、13.2、15.5、15.9、17.4、17.8、20.2、21.7、22.1、22.8、23.4和24.3;5.4、7.9、9.5、11.9、18.1、18.6、20.9、30.2和32.1。cn105616362a公开了其药物组合物中帕瑞昔布钠为结晶形式,使用cu-kα射线进行x射线粉末测定,所示衍射角2θ为6.1912、8.3750、10.9145、12.1060、14.5531、17.0537、18.0569、20.5835、22.7235、23.9818、25.5648、29.9764。cn104910091a,公开了的晶型在2θ为2.58、9.57、11.05、14.59和16.54处有峰。cn104418818a,公开的无水化合物晶体具有下列2θ衍射角:4.50、7.28、9.13、12.62、12.71、13.50、14.55、14.92、15.12、16.44、16.50、17.15、18.34、19.87、20.10、20.34、20.67、22.50、23.91。cn104418819a,公开了一种半水合物晶体,其图谱具有下列2θ衍射角:10.120、11.300、12.180、14.480、19.060、21.470、24.320、26.460、27.140、28.220、28.940、29.580、30.940、32.960、33.520、34.840、36.450、38.000、39.340、41.160。cn105125506a说明书0044验证试验3提及原研市售品及cn105125506a冻干样品晶型测试结果均为无定型、现有技术中预冻采用一步快速降温后的冻干样品晶型检测峰值为5.004、11.440、22.339、31.060、31.900、32.800。现有技术的帕瑞昔布钠存在一定的缺陷,尤其是其具有引湿性,容易吸潮导致水解为伐地昔布,不利于用药安全。在这种情况下,开发具有更多优越性能的帕瑞昔布钠的晶型很有必要。本发明研究人员做了大量实验,考察了多种溶媒和水,及其之间不同的比例,考察了多种干燥条件和结晶条件,在经过不断地研究改进后,发明并制备了本发明的帕瑞昔布钠晶体化合物,其性状为白色结晶性粉末,晶粒大小均匀,流动性好。该晶体化合物在常温干燥条件下不会发生结晶水的流失,在高湿状态下不易吸湿,贮存稳定性好。技术实现要素:本发明的第一目的是提供一种帕瑞昔布钠晶体化合物,所述帕瑞昔布钠晶体化合物稳定性好,即使在高湿状态下也不易吸湿。为了实现该目的,采用的技术方案是:一种帕瑞昔布钠晶体化合物,该晶体化合物为帕瑞昔布钠一水合物,其分子式为:c19h17n2nao4s·h2o,以2θ±0.2°衍射角表示的x-射线粉末衍射图谱在2.055°、3.045°、3.727°、9.849°、11.636°、14.209°、27.418°、28.636°、30.582°、31.418°、32.436°、34.018°、35.782°、36.927°、38.618°处显示有特征衍射峰,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示。该晶体化合物熔点为265~268℃,无引湿性,在25℃暴露于95%rh时,引湿增重小于0.1%。研究表明,在x-射线粉末衍射图谱中,由新晶型得到的衍射谱图对于特定的晶型往往是特征性的,其中谱带(尤其是在低角度)的相对强度可能会因为结晶条件、粒径和其它测定条件的差异而产生的优势取向效果而变化。因此,衍射峰的相对强度对所针对的晶型并非是特征性的,判断是否与已知的晶型相同时,更应该注意的是峰的相对位置而不是它们的相对强度。经试验证实,本发明所提供的帕瑞昔布钠晶体化合物含有1个结晶水,且其粉末x-射线衍射图谱与现有技术具有明显不同的峰的相对位置,可见其是一种与现有技术不同的新晶型。本发明的第二目的是提供一种帕瑞昔布钠晶体化合物的制备方法。为了实现该目的,采用的技术方案是:一种帕瑞昔布钠晶体化合物的制备方法,具体包括如下步骤:(1)将帕瑞昔布钠粗品加入到有机溶剂1中,加热至40~50℃;(2)粗品溶解后加入活性炭,保持40~50℃状态下脱碳20~50min,过滤;(3)将步骤(2)所得滤液转入三口瓶中,加入有机溶剂2,转入冷肼中,控温-5~5℃搅拌析晶3~5h;(4)抽滤,少量有机溶剂2洗涤,得结晶性固体,45~55℃下真空干燥6~10h,得帕瑞昔布钠一水合物。优选地,有机溶剂1为n,n-二甲基甲酰胺和纯化水的混合溶液;有机溶剂2为乙酸乙酯。优选地,有机溶剂1中n,n-二甲基甲酰胺与纯化水的体积比为4.0~6.0:1.0。优选地,步骤(1)中帕瑞昔布钠粗品与n,n-二甲基甲酰胺的质量体积比为1g:4~6ml;步骤(3)中帕瑞昔布钠粗品与乙酸乙酯的质量体积比为1g:15~25ml;步骤(4)中帕瑞昔布钠粗品与乙酸乙酯的质量体积比为1g:1~3ml。优选地,步骤(1)中帕瑞昔布钠粗品与n,n-二甲基甲酰胺的质量体积比为1g:5ml;步骤(3)中帕瑞昔布钠粗品与乙酸乙酯的质量体积比为1g:20ml;步骤(4)中帕瑞昔布钠粗品与乙酸乙酯的质量体积比为1g:2ml。下面通过对本发明提供的帕瑞昔布钠晶体化合物进行研究来进一步解释和说明本发明技术方案:1、元素分析c19h17n2nao4s·h2o采用美国perkin-elmer公司pe2400ⅱ元素分析仪对本发明制备得到的帕瑞昔布钠晶体化合物进行元素分析:元素分析(%)理论值为:h(4.6662),c(55.6025),n(6.8256),o(19.4915),s(7.8127);元素分析(%)测定值为:h(4.6681),c(55.6042),n(6.8239),o(19.4923),s(7.8107);结果显示:元素分析测定值与元素分析理论值基本相符。2、晶型检测取本发明制备得到的帕瑞昔布钠晶体化合物,使用cu-kα射线测量得到的x-射线粉末衍射图如图1所示,其以2θ±0.2衍射角表示的x-射线粉末衍射图谱在2.055°、3.045°、3.727°、9.849°、11.636°、14.209°、27.418°、28.636°、30.582°、31.418°、32.436°、34.018°、35.782°、36.927°、38.618°处显示有特征衍射峰。3、水分分析采用卡式水分测定仪测定,本发明的帕瑞昔布钠晶体化合物的水含量为4.396%,与理论值4.389%相符。4、热重分析(tga)与差热分析(dsc)取本发明制备得到的帕瑞昔布钠晶体化合物,使用tgaq500热分析仪进行热重分析(tga),结果显示本发明结晶在115℃左右快速失去约一个水分子的重量,并在100℃之前无明显重量变化,证实其失去的水分子为结晶水分子,而非游离水分子;使用pyris-1差示扫描量热仪进行差热分析(dsc),得图2。5、纯度检测经hplc纯度检测,本发明制备得到的帕瑞昔布钠晶体化合物的纯度可达到99.96~99.99%。6、熔点检测取本发明制备得到的帕瑞昔布钠晶体化合物进行检测,熔点为265~268℃。与现有技术相比,本发明具有如下优点:(1)本发明所提供的帕瑞昔布钠晶体化合物是一水合物结晶,是一种不同于现有技术的新的水合物晶型;(2)本发明所提供的帕瑞昔布钠晶体化合物改善了帕瑞昔布钠原料的引湿性,即使在高湿状态下也不易吸湿,容易控制其物理特性;(3)本发明所提供的帕瑞昔布钠晶体化合物稳定性好、室温下能够较长时间保存,且流动性好,非常适合临床应用;(4)本发明所提供的帕瑞昔布钠晶体化合物制备方法操作简单,适合大规模生产。附图说明图1为本发明制备得到的帕瑞昔布钠晶体化合物的x-射线粉末衍射图;图2为本发明制备得到的帕瑞昔布钠晶体化合物的tga-dsc图。具体实施例实施例将有助于进一步了解本发明的技术方案的优点、效果,不限定本发明的保护范围,本发明的保护范围由权利要求来决定。对本领域技术人员显而易见的是,对于材料和方法两者的许多改变可在不脱离本发明范围的情况下实施。实施例1、本发明晶体化合物的制备将帕瑞昔布钠粗品100.00g加入到500mln,n-二甲基甲酰胺和100ml纯化水组成的混合溶剂中,加热至45℃,溶解;固体溶解后,加入5g活性炭,保持45℃状态下脱碳30min,过滤;将滤液转入三口瓶中,加入乙酸乙酯2000ml,转入冷肼中,控温0℃搅拌析晶4h;抽滤,200ml乙酸乙酯洗涤,得结晶性固体,50℃下真空干燥8h,得帕瑞昔布钠一水合物89.98g。实施例2-12:下面通过实验例进一步说明本发明:实验例1、引湿性试验本实验例考察了本发明提供的帕瑞昔布钠晶体化合物的引湿性,按照中国药典2010版二部附录ⅺⅹj药物引湿性试验指导原则进行,设定人工气候箱的温度为25℃,相对湿度为95%。表1:引湿性试验结果样品12345678引湿增重百分率0.0810.0880.0860.0870.0915.356.780.397样品1-5为实施例1制备工艺制备的5批样品;样品6-7为市售帕瑞昔布钠原料的样品;样品8参照专利cn104418819a制备的半水合物;从表1可以看出,本发明帕瑞昔布钠晶体化合物无引湿性,在25℃暴露于95%rh时,引湿增重小于0.1%。与市售的帕瑞昔布钠原料药相比,引湿性明显改善;与专利cn104418819a制备的半水合物相比,引湿性也有较大改善。实验例2:流动性实验本实验例采用固定漏斗法测定各样品的休止角,从而评价本发明提供的帕瑞昔布钠晶体化合物的流动性。具体方法如下:将漏斗置于坐标纸上的适宜高度,取实施例1制备的样品6批,从固定的漏斗中自由流下,直到形成的圆锥体顶部与漏斗口接触,测算物料堆积层的斜边与水平线的夹角度数(休止角θ)。实验结果如表2所示。表2:流动性实验结果样品123456平均值θ(°)32.5332.6133.2833.4333.5233.7433.185从表2的实验结果分析,本发明实施例1制备得到的帕瑞昔布钠晶体化合物的流动性很好,有利于提高分装的准确性,并且与其他成分混合时易于混合均匀。对本发明其他实施例制备的样品也进行了测定,得到了相似的实验结果。实验例3:杂质检测分析结果本实验例对实施例1制备的5批样品中的有关物质杂质进行检测分析,按照中国药典2010版第二部附录ⅺⅹf药品杂质分析指导原则进行,结果见表3。表3:各样品杂质检测分析结果样品总杂(%)其他单杂(%)伐地昔布(%)含量(%)10.0930.0310.02399.9020.0910.0300.02699.9130.0920.0350.02599.8840.0930.0340.02799.8950.0920.0350.02699.85对本发明其他实施例制备的样品也进行了检测,得到了相似的实验结果。实验例4、稳定性试验本实验例通过加速试验和长期试验,考察本发明提供的帕瑞昔布钠晶体化合物的化学稳定性。1、加速试验方法:根据中国药典规定,将本发明实施例1制备的5批样品模拟上市包装置于40℃±2℃,相对湿度75%±5%条件下放置6个月,分别于1、2、3、6个月末取样分析,并与0天样品检测结果比较。表4:加速试验结果结论:试验结果表明,本品经加速试验6个月,各项考察项目检测指标均无明显变化,各样品稳定性较好。性状在0至6个月考察期间,各时间点均符合规定。对本发明其他实施例的样品也做了加速试验,得到了相似的试验结果。2、长期试验方法:根据中国药典的规定,将本发明实施例1制备的5批样品模拟上市包装置于25℃±2℃,相对湿度60%±5%条件下放置24个月,分别于0、3、6、9、12、18、24个月末取样分析,并与0天样品检测结果比较。表5:长期试验结果结论:本品经长期试验24个月考察,各项考察项目检测结果与放置前比较均无明显变化,各样品稳定性较好。对其他实施例的样品也做了长期试验,得到了相似的试验结果。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1