EOC315Mod.I晶型化合物及其制备方法与流程
本发明属于医药
技术领域:
,具体涉及一种eoc315mod.i晶型化合物的制备及表征,还涉及所述的eoc315mod.i晶型物在制药中的应用。
背景技术:
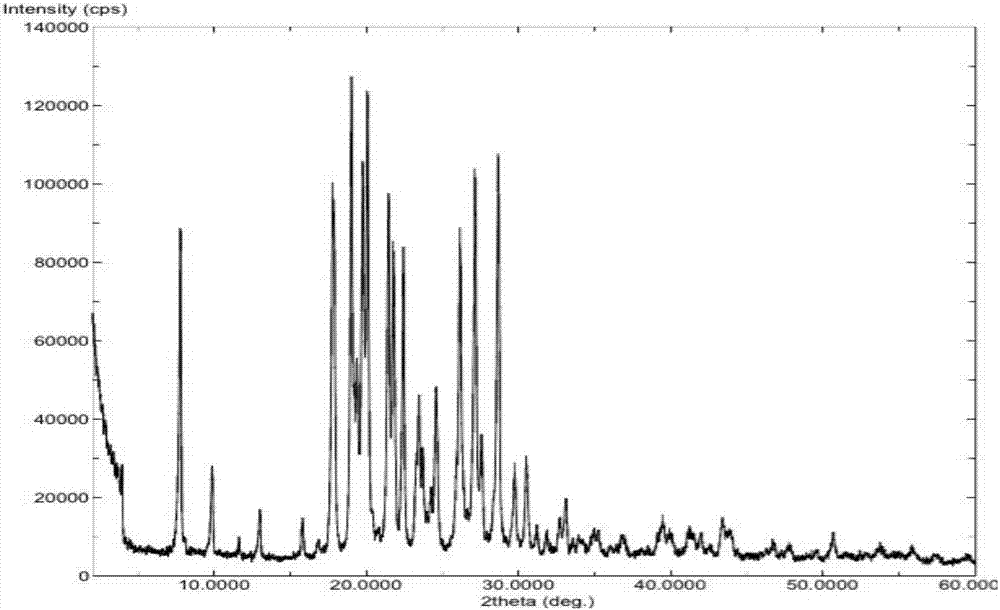
:eoc315(4-(4-氯苯胺基)-7-(2-甲氨基羰基-4-氧甲基)吡啶基呋喃并[2,3-d]哒嗪甲磺酸盐)目前尚未在任何国家上市。美国生物制药企业actbiotech,inc.(actbiotech,以下简称act)于2008年从bayerhealthcarepharmaceuticals获得了eoc315的许可,2014年1月,亿腾与act公司签订了资产购买协议,获得其全球独家权利。其结构式如下所示:eoc315是一种具有口服生物利用度的血管内皮生长因子受体2(vegfr2)酪氨酸激酶活性的强效抑制剂,根据生化测定的测量结果显示,ic50值为6nm。该药还抑制血小板源生长因子受体(pdgfr)酪氨酸激酶活性,其中ic50值为15nm。这2种受体在涉及刺激内皮细胞形成血管的血管生成过程和表达pdgfr的周皮细胞支撑新形成血管的过程中发挥着关键作用。eoc315是一种正处于开发阶段的选择性最强的已知vegfr抑制剂,其能够将上述靶点与其他激酶区分开来。作为一种口服制剂,患者能够实现对预期靶点进行持续、长期的抑制。eoc315在国际上处于ii期临床开发阶段,作为与卡培他滨/顺铂联用的抗癌药物用于晚期未经治疗的胃癌的化疗。迄今为止,eoc315已经累积在超过300例患者,进行了共七项i、ib期和ii期临床研究。多项临床研究探索了eoc315在多种晚期实体瘤患者中的安全性和有效性。结果显示eoc315(单药和联合治疗)具有良好的耐受性,并显示出良好的抗肿瘤前景。特别是,ii期临床研究eoc315在晚期胃癌或胃-食管结合部(gej)癌症患者中评价了eoc315与卡培他滨和顺铂联用作为一线治疗的有效性和安全性。结果显示,其总缓解率(orr)达到67%,几乎是单独化疗所得历史缓解率的两倍。中位无进展生存期(pfs)为4.5个月。kaplan-meier分析显示6个月内无进展患者比例为52%。出现肿瘤缓解的患者的中位进展时间为6.4个月。因此,目前的临床开发拟用于治疗胃癌或胃-食管结合部癌症的治疗。美国拜尔公司在其专利cn00816369和cn200510127109中,公开了化合物eoc315及其类似物的合成方法,并同时公开其药物组合物及药物组合物在药学中的应用。本公司在zl200510140054中亦公开了一种eoc315的适于工业化生产的制备方法。迄今为止,还没有查询到eoc315晶型相关的公开报道,本发明首次公开用于药学研究的eoc315的mod.i晶型。技术实现要素:本发明的目的之一在于提供一种稳定eoc315mod.i晶型并对其进行表征。本发明另一目的在于提供所述晶型的制备方法。本发明的另一目的在于提供eoc315微粉化前后x-粉末衍射峰值相对强度,dsc及结晶形状的变化。本发明的另一目的在于说明eoc315微粉化不会改变其晶型,仍为mod.i晶型。本发明进一步的目的在于提供一种包含eoc315mod.i的药物组合物。本发明的其他目的还在于提供所述eoc315mod.i晶型的药物在vegfr-2(血管内皮生长因子受体2)和pdgfr(人血小板源性生长因子受体)激酶活性抑制剂中的应用。本发明的技术方案如下:本发明的如式i所示的eoc315mod.i晶型化合物:式1当使用x射线衍射cukα分析时,x-射线粉末衍射图中在4.01,7.85,9.94,13.04,19.08,19.46,20.10,21.82,22.49,23.76,24.26,27.17,28.52,30.48处出现x射线衍射峰。x射线衍射图的2θ、d(a)见表1。表1.本发明所述eoc315mod.i晶型2θ和d(a)值2θd(a)2θd(a)2θd(a)4.00622.0424.2653.66537.042.4257.84911.25524.6793.60437.502.3969.9368.89526.2013.39838.5192.33511.7187.54627.173.28039.302.29115.8615.58327.613.22840.002.25217.7914.98128.6583.11241.302.18419.0844.64729.8282.99342.432.12919.4634.55730.5192.92343.392.08419.8064.47931.8712.80546.771.94120.104.41432.672.73847.711.90520.4814.33333.132.70249.531.83921.8184.07033.8692.64550.671.80022.4863.95133.6412.66253.501.71123.7573.74234.7462.579当使用x射线衍射cukα分析时,所述x射线衍射图如图1所示。mod.i晶型化合物熔点为200.6℃,dsc热分析结果如图3;tga分析结果显示,mod.i晶型化合物降解温度为241.71℃,其tga图加图8。mod.i晶型化合物红外光谱(kbr)在3415cm-1、3058cm-1、2805cm-1、1668cm-1、1652cm-1和1227cm-1处有谱带,其红外检测结果如附图7。mod.i晶型化合物热台偏光显微镜分析结果显示该产品为针状结晶,颗粒较大(长度可达几百微米),结晶度高。其hsm图如图5。本发明还提供了所述的mod.i晶型化合物的制备方法,该方法包括:eoc315自由碱溶解在有机溶剂中,逐滴加入甲磺酸的有机溶液,滴完后蒸出部分溶剂,然后加入另一溶剂降温结晶得到eoc315mod.i晶型化合物。mod.i晶型化合物的制备方法,详细制备方法如下:1)eoc315的自由碱溶于溶剂中制成eoc315溶液,甲磺酸溶于溶剂中制成甲磺酸溶液;2)在20~90℃将甲磺酸溶液加入eoc315溶液中;3)减压蒸馏除去大部分溶剂,加入极性或者非极性溶剂,0~30℃打浆,过滤,收集滤饼;4)25~90℃真空干燥。所述的步骤1)中,溶剂为甲醇、乙醇、异丙醇、丁醇、己烷、庚烷、丙酮、乙丙醚、四氢呋喃、甲苯、乙酸乙酯和乙腈的单个有机溶剂或者其混合溶剂。优选地,所述的步骤1)中,溶剂为甲醇、乙醇、异丙醇和丁醇的单种溶剂或者其混合溶剂。药物的颗粒大小影响该药物在各种溶剂中的溶解度,特别是对口服固体制剂,直接影响该药物在人体中的生物利用度,生物利用度的大小直接决定药物的药效。将干燥后得到的eoc315mod.i型晶体微粉化,粒径d90≤15μm,得到eoc315mod.i晶型化合物微粉。eoc315mod.i晶型化合物微粉,当使用x-射线粉末衍射cukα分析时,x-射线衍射图如图2所示,和微粉化前图谱基本一致,说明微粉化前后样品晶型没有发生变化,但是固体性质发生变化,例如颗粒的大小及择优定向可能会影响图谱的峰值相对强度,但是不会影响其峰值的位置特别是低度峰值的位置,微粉化后的图谱也显示一定的基线隆起,表明可能会有一定的结晶度的问题。微粉化后x射线衍射图的2θ、d(a)见表2。表2.eoc315mod.i晶型微粉化后的2θ和d(a)值2θd(a)2θd(a)2θd(a)3.89822.6523.4133.79635.2092.5477.76611.37523.6163.76436.742.4449.8568.96724.233.67039.382.28612.9536.82924.5213.62739.922.25615.7775.61326.093.41341.112.19417.7984.97927.073.29141.462.17618.9984.66727.5343.23741.912.15419.3574.58228.6083.11843.342.08619.7344.47529.7153.00446.7081.94320.0394.42730.4802.93047.8381.89921.4234.14431.802.81250.611.80221.7424.08432.662.74022.3943.96733.052.70823.273.82033.8692.645eoc315mod.i晶型化合物微粉,其转变温度为196.39℃,低于微粉化前的200.6℃,说明微粉化后结晶度较低,微粉化后dsc热分析分析结果如图4。eoc315mod.i晶型化合物微粉,其热台偏光显微镜(hsm)分析结果显示该样品为微粉结晶(小于10微米),结晶度较低,其hsm分析结果如图6。eoc315mod.i晶型化合物微粉,根据其hsm结果,x-粉末衍射和dsc结果显示为mod.i晶型化合物。eoc315mod.i晶型化合物微粉与微粉化前的晶型未发生变化,均为mod.i晶型化合物,只是微粉化前颗粒度较大,洁净度较高。另一方面,本发明还提供了一种药物组合物,该药物组合物包含:本发明所述的eoc315mod.i晶型化合物,以及一种或多种药学上可接受的载体或赋形剂。本发明的eoc315mod.i晶体可以与一种或多种药学上可接受的载体或赋形剂制成药学领域常用剂型,如片剂、胶囊剂、注射剂等制剂,优选固体口服制剂。用于口服给药的药物组合物可以通过eoc315mod.i型晶体化合物和一种或多种固体载体混合得到,如果需要的话将所得混合物造粒,并进行颗粒的混合,如果需要或有必要,通过加入额外的赋形剂的方式,从而得到片剂或片芯。特别地,合适的载体是填充剂(例如糖,如乳糖、蔗糖、甘露醇或山梨醇、纤维素制剂和/或磷酸钙,如磷酸三钙或磷酸氢钙),以及粘合剂(例如淀粉,如玉米淀粉、小麦淀粉、大米淀粉或土豆淀粉,甲基纤维素,羟丙基甲基纤维素,羧甲基纤维素钠,和/或聚乙烯吡咯烷酮),如果需要的话,和/或崩解剂(例如上述的淀粉,以及羧甲基淀粉、交联聚乙烯吡咯烷酮、藻酸或其盐,如藻酸钠)。特别地,额外的赋形剂是流动调节剂和润滑剂,例如硅酸、滑石、硬脂酸或其盐,如硬脂酸镁或钙,和/或聚乙二醇或其衍生物。片芯可以有合适的包衣,所述包衣通过使用特别是浓糖溶液(其可以包含阿拉伯胶、滑石、聚乙烯吡咯烷酮、聚乙二醇和/或二氧化钛)、或在合适的有机溶剂或溶剂混合物中的包衣溶液进行。可以向所述片剂或片剂包衣中加入染料或者颜料,例如出于识别目的或者为了显示不同剂量eoc315。另一方面,本发明还提供了所述的eoc315mod.i型晶体化合物是一种强效的vegfr-2(血管内皮生长因子受体2)和pdgfr(人血小板源性生长因子受体)激酶活性抑制剂,可以有效抑制相关肿瘤细胞的生长。eoc315可以与标准治疗药物紫杉醇卡培他滨和吉西他滨联合用药,1)与单独给药任一种药剂相比,产生更好的减少肿瘤生长或甚至清除所述肿瘤的效力;2)提供被给药的化疗剂的更少给药量;3)提供化疗方法,该方法患者耐受良好,有害的药理学并发症比单一药剂化疗和某些其他联合疗法所观察到的少;3)提供更宽广的哺乳动物尤其是人类的不同癌症类型的治疗谱;4)提供更高的治疗患者的响应速度;5)与标准的化疗相比提供更长的治疗患者的生存时间;6)与其他癌症药剂联合产生拮抗作用的已知情况相比,提供更长的肿瘤进展时间,和/或产生至少和单独使用所述药剂一样的效力和耐受性。可以药学有效量和这些细胞毒素剂中的一种或多种同时地、分开地或连续地给药。给药的联合的活性剂的量(“药学有效量”)在很广的范围内变化,并取决于待治疗的病症和给药方式。所述量可以涵盖对预期的治疗有效的任意量。决定联合的活性剂的“药学有效量”是在本领域技术人员的能力范围之内的。附图说明图1为eoc315mod.i晶型x-射线粉末衍射图;图2为eoc315mod.i晶型微粉化后的x-射线衍射图;图3为eoc315mod.i晶型差示扫描量热(dsc)图;图4为eoc315mod.i晶型微粉化后差示扫描量热(dsc)图;图5为eoc315mod.i晶型热台偏光显微镜(hsm)结果显示图;图6为eoc315mod.i晶型微粉化后热台偏光显微镜(hsm)结果显示图;图7为eoc315mod.i晶型红外光谱(ir)图;图8为eoc315mod.i晶型的热重分析(tg/dta)图。具体实施方式通过下面的实施例可以对本发明进一步的描述,然而,本发明的发明并不限于下面的实施例,这些实施例不以任何方式限制本发明的范围。本领域的技术人员在权利要求的范围内所作出的某些改变和调整也应认为属于本发明的范围。实施例1eoc315mod.i晶型化合物的制备eoc315mod.i型晶体化合物的制备包括如下步骤:1)9geoc315碱溶于90ml甲醇中制成eoc315溶液,2.5g甲磺酸溶于16ml甲醇中制成甲磺酸溶液;2)在50℃时开始,将甲磺酸溶液滴加入eoc315溶液中,滴完之后继续加热15分钟;3)对滤液减压蒸馏,蒸出部分溶剂后加入200ml异丙醇继续减压蒸馏,直至料液约为250ml,降温析晶1小时,抽滤,收集滤饼。4)45℃真空干燥得到eoc315mod.i晶型化合物,收率90.5~96.3%,纯度99.1~99.9%。使用x射线衍射分析得到如图1所示的x-射线粉末衍射图。实施例2用差示扫描量热法(dsc)分析实施例1的eoc315mod.i晶型化合物在氮气氛下以10℃/min的加热速度在铝盘中记录dsc扫迹,测得通过实施例1所得eoc315在200.7℃有一尖锐吸热峰,说明eoc315为单一mod.i晶型,熔点为200.7℃。分析分析结果如图3。本领域普通技术人员知晓,测定的熔解温度取决于实验条件,尤其是所用的加热速度。另外,熔解温度受到物质纯度的影响。报道的熔解温度是用纯度至少98.5%的产品测定的。实施例3用x粉末衍射分析实施例1的eoc315mod.i晶型化合物在室温下,用锗致单色化cukα1-辐射记录x-射线粉末衍射数据。在室温条件下,使用小线型位置敏感检测仪在3°≤2θ≤35°(步长0.5°)之间用0.08°的角分辨率进行2θ扫描。x-射线粉末衍射图如图1,其中2θ与d(a)值如表1所示。本领域普通技术人员会明了获得的x-射线衍射图谱可能存在测量误差,该误差取决于测量条件。特别地,通常已知x-射线衍射图谱的强度可能根据物质的晶癖和使用的测量条件而波动。另外,在给定温度下常规的x-射线衍射图谱的衍射角θ的测量误差通常约为±0.1°,并且在涉及到衍射角时应当将这种程度的测量误差考虑在内。因此,具有和在附图中公开的x-射线粉末衍射图谱基本一致的x-射线衍射图谱的任何晶型都落入本发明的范围之内。实施例4实施例1的eoc315mod.i晶型化合物的热重分析(tg/dta)在氮气氛下以10℃/min的加热速度,测得通过实施例1所得eoc315在188.9℃质量分数为99.8%,失重0.2%,基本无失重,表明样品中不含结晶溶剂,挥发性物质的含量也很低。供试品dta曲线在198.1℃有一吸热峰,与dsc结果一致,其tga图如图8。本领域普通技术人员知晓,测定的温度取决于实验条件,尤其是所用的加热速度。另外,温度受到物质纯度的影响。报道的熔解温度是用纯度至少98.5%的批量测定的。实施例5eoc315mod.i晶型的热台偏光显微镜(hsm)分析对于eoc315样品,hsm分析结果显示该样品为针状结晶,粒度较大(长度可达几百微米),结晶度高,其hsm图见附图5。实施例6实施例1的eoc315mod.i晶型化合物的的红外分析使用漫反射(kbr),记录eoc315mod.i型晶体红外光谱。红外光谱图见图7,主要的红外波段及其归属见表3。表3.eoc315mod.i型晶体红外光谱主要吸收峰及其归属本领域普通技术人员知晓,基于使用的仪器和测量条件,可接受的波数位移容限是±2cm-1。因此,具有和在附图中公开的ft-拉曼光谱基本一致的ft-拉曼光谱的任何固态形式都落入本发明的范围之内。实施例7将实施例1所得eoc315mod.i晶型化合物微粉化设定微粉设备压力应≥0.65mpa,微粉化后得到eoc315晶型化合物微粉,粒径d90≤15μm。利用上述方法,测得微粉化后样品的x-粉末衍射,dsc,及hsm,其结果见图2、图4和图6。从图示结果可以看出,微粉化前后eoc315晶型未发生变化,仍为mod.i晶型。实施例8eoc315mod.i晶型化合物对肿瘤细胞的抑制作用eoc315mod.i晶型化合物在体外显示生化和细胞活性,与抗血管生成的作用机制一致。因此,在colo-205人体crc异种移植模型中,实施了一项研究以评估体内观察到的抗肿瘤活性是否与这一假设一致。对内皮细胞标志物cd31染色后,通过定量组织形态测定法,检查了肿瘤的微血管密度。药物首次给药后4小时,与各溶媒对照组相比,肿瘤微血管面积(mva)明显减小(32%;p<0.05)。24小时后,肿瘤mva的下降水平进一步增加(53%;p<0.01)。用药3天后,效果更佳明显,并且在4小时和24小时观察到mva分别下降64%和68%。根据上述实验结果,本领域技术人员可以发现,如果使用eoc315mod.i晶型化合物,可以明显抑制肿瘤细胞的生长。实施例9在晚期胃癌或胃食管接合部(gej)癌症患者中考察eoc315与卡培他滨(x)和顺铂(p)联用研究在最多6个周期的顺铂治疗后,根据毒性反应情况,受试者以既往剂量继续使用卡培他滨和eoc315或者使用卡培他滨或eoc315进行单药治疗,直至疾病进展。该研究共入组48例受试者。其中的45例受试者(94%)存在肿瘤转移,35例受试者(73%)有两个以上转移部位,26例受试者(54%)有肝累及。26例受试者(54%)的主要累及部位为胃食管接合部,22例受试者(46%)为胃部,大部分受试者为ecog1分(n=41,85%)。39例受试者至少完成了1个疗程的治疗并进行了疗效评估。客观缓解率(orr)为67%。1例(2.6%)受试者达到了完全缓解,25例(64.1%)受试者达到了部分缓解。11例(28.2%)受试者病情稳定并维持12周以上的时间。评估显示客观缓解一般发生在治疗的前两个周期,持续时间较长,与受试者接受治疗的时间长度密切相关。抗肿瘤效果与肿瘤的组织类型、原发部位、肝脏是否受累及研究中心无关。中位无进展生存期(pfs)为4.5个月。kaplan-meier分析显示6个月内无进展患者比例为52%。出现肿瘤缓解的患者的中位进展时间为6.4个月。中位os估计为7.8个月,1年生产率为33%。初步结果提示总体缓解率较高(67%的客观缓解率(orr),几乎是单独化疗所得历史缓解率的两倍),缓解时间较长,有证据提示生存期延长。实施例10稳定性实验结果在25℃,相对湿度60%,12个月的长期稳定性或者40℃,相对湿度75%,6个月的加速稳定性考察,结果如下表4和表5:表4eoc315长期稳定性试验(温度25℃±2℃,相对湿度rh60%±5%)表5.eoc315加速条件下稳定性试验(40℃±2℃、相对湿度75%±5%)从上表可以看出,eoc315的mod.i晶型为热力学上的稳定结构。为了提高其生物利用度,对生产出来的药物微粉化,微粉化后的药物晶型仍为mod.i晶型,只是其结晶度变低。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1