3‑叔丁基‑4‑羟基苯丙酸的制备方法与流程
本发明涉及一种3-叔丁基-4-羟基苯丙酸的制备方法。
背景技术:
紫外线吸收剂是一种光稳定剂,它能吸收阳光及荧光光源中的紫外线部分,自身却不发生变化,被广泛应用于塑料、涂料、染料、汽车挡风玻璃、化妆品、防晒剂等行业。uv384是其中的代表产品,因其具有与合成材料相容性好、挥发性低、毒性小等优点,使其具有较大的市场需求量。
3-叔丁基-4-羟基苯丙酸是合成紫外线吸收剂uv384的重要中间体,随着uv384市场需求量的增加,3-叔丁基-4-羟基苯丙酸的需求量也逐渐增加。目前,工业上3-叔丁基-4-羟基苯丙酸的生产工艺是以3,5-二叔丁基-4-羟基苯丙酸甲酯为原料,经过脱烷基化得到中间体3-叔丁基-4-羟基苯丙酸甲酯,采用重结晶的方法对中间体进行提纯后在碱性条件下水解得到产品3-叔丁基-4-羟基苯丙酸钠盐,酸化后得到产品。该工艺具体反应方程式如下所示:
该工艺存在的问题是在脱烷基化反应过程中,当原料的转化率达到一定程度时(一般当原料转化率达到70%左右副产物开始明显增加),会产生多种副产物,包括脱掉两个叔丁基的对羟基苯丙酸甲酯(杂质1)、脱掉丙酸酯基的2,6-二叔丁基苯酚(杂质3)及中间体之间的酯化产物(杂质2)等(其中以中间体之间的酯化产物2为主要副产物)。如果要保证产品质量,必须对中间体进行分离纯化,因而增加了工艺成本,降低了产品的总收率(收率仅为60%左右)。另外,脱烷基化反应中产生的副产物不能回收利用,只能作为废弃物排放,增加环境污染治理的负担。因此,如何降低副产物的量,提高产品的总收率,成为需要解决的问题。
技术实现要素:
本发明是要解决现有3-叔丁基-4-羟基苯丙酸制备过程中反应选择性差、收率低、环境污染严重的问题,提供一种3-叔丁基-4-羟基苯丙酸的制备方法。
本发明3-叔丁基-4-羟基苯丙酸的制备方法,包括以下步骤:
一、脱烷基化反应:将3,5-二叔丁基-4-羟基苯丙酸甲酯与催化剂加入反应器中,加热升温至60~90℃,hplc监控反应,当中间体质量百分含量达到65%~80%时停止反应;其中催化剂的质量为3,5-二叔丁基-4-羟基苯丙酸甲酯质量的1.5%~3%;所述中间体为3-叔丁基-4-羟基苯丙酸甲酯;
二、选择性水解:向步骤一所述的反应器中加入氢氧化锂水溶液,在60~80℃条件下反应至体系呈澄清状态,然后用稀盐酸调节水解体系ph为6.5~7.5,向体系中加入有机溶剂萃取未反应的原料,得到水相和有机相,将有机相回收溶剂得到有机相浓缩物;
三、分离提纯:将步骤二得到的水相用稀盐酸酸化至ph为1-2,析出白色固体,过滤,得3-叔丁基-4-羟基苯丙酸粗品;
将3-叔丁基-4-羟基苯丙酸粗品用乙腈-正丁醇混合液重结晶,得到的白色固体部分是纯品3-叔丁基-4-羟基苯丙酸,将重结晶的母液回收溶剂得到母液回收物备用;
四、回收利用:
将步骤二得到的有机相浓缩物与催化剂加入反应器中,加热升温至60~90℃,hplc监控反应,当中间体质量百分含量达到65%~80%时停止反应;其中催化剂的质量为步骤二得到的有机相浓缩物质量的1.5%~3%;所述中间体为3-叔丁基-4-羟基苯丙酸甲酯;
然后向反应器中加入氢氧化锂水溶液,在60~80℃条件下反应至体系呈澄清状态,然后加入步骤三重结晶的母液回收物(作为步骤二反应物的中和剂),再用稀盐酸调节水解体系ph为6.5~7.5,向体系中加入有机溶剂萃取,得到水相;
将水相用稀盐酸酸化至ph为1-2,析出白色固体,过滤,得3-叔丁基-4-羟基苯丙酸粗品;将3-叔丁基-4-羟基苯丙酸粗品用乙腈-正丁醇混合液重结晶,得到的白色固体部分是纯品3-叔丁基-4-羟基苯丙酸;
五、收集步骤三和步骤四得到的纯品3-叔丁基-4-羟基苯丙酸,即制备过程完成。
进一步的,步骤一和步骤四中催化剂为路易斯酸,所述路易斯酸为对甲苯磺酸、甲基磺酸、磷酸或硫酸。
进一步的,步骤二和步骤四中氢氧化锂水溶液的质量百分浓度为10%-20%,其中氢氧化锂与3,5-二叔丁基-4-羟基苯丙酸甲酯的摩尔比为(2.0-2.2)∶1。
进一步的,步骤二和步骤四中萃取所用的有机溶剂为乙酸乙酯、甲苯、正丁醇、石油醚、二氯甲烷或正己烷。
进一步的,步骤二中有机溶剂的体积与原料3,5-二叔丁基-4-羟基苯丙酸甲酯的质量比为(0.5~5.0)ml:1g。
进一步的,步骤三中乙腈-正丁醇混合液中乙腈与正丁醇的质量比为(0.6~1.0)∶1,3-叔丁基-4-羟基苯丙酸粗品的质量与乙腈-正丁醇混合液的体积比为1g:(1.0-2.5)ml。
上述方法中所述稀盐酸的浓度均为3mol/l。
现有的方法中原料结构中存在两种烷基,即叔丁基和羧丙基,这样导致这两种烷基在相同脱烷基化反应条件下都会发生脱去反应,当脱除羧丙基时即可生成杂质3,脱除两分子叔丁基时生成杂质1;同时脱烷基化反应的酸性条件也是酯交换的反应条件,当中间体的含量过高时,就会发生两分子中间体的酯交换反应,生成杂质2,因此现有方法的反应选择性较差。
本发明的原理如下:
本发明的关键是采用了一种使中间体可以选择性水解的方法,即以lioh水溶液为水解剂,在温和的条件下选择性的将中间体3-叔丁基-4-羟基苯丙酸甲酯水解得到相应的羧酸,而原料3,5-二叔丁基-4-羟基苯丙酸甲酯和中间体酯化产物(杂质2)中的酯基不受影响。
水解反应结束后,采用两次中和的方法达到中间体与原料及主要副产物的分离。其具体原理为:首先将水解反应液中和至ph7左右,此时各种组分中的酚羟基被游离出来,使原料和主要副产物成为中性分子而在有机溶剂萃取时进入有机相,中间体水解产物则以羧酸锂盐的形式进入水相。分出有机相后,水相中的锂盐经第二次酸化至ph为1~2,使其转化成羧酸,得到的产品直接从水中析出,如此获得的粗产品纯度可以达到95%~97%。
由于本发明方法中未反应的原料可以回收利用,所以脱烷基化反应不需要达到完全,可以在副产物较少时停止反应而提高反应的选择性。本发明的实际操作中一般控制主要副产物在3%以下(如果转化率达到90%以上时,该副产物的含量可达15%以上),原料转化率80%左右,中间体含量为65%-80%时停止反应,如此处理得到的萃取液脱除溶剂后的组成大致为:原料70%-85%,主要副产物10%-20%,这样的浓缩液可以作为原料进行脱烷基化反应。
粗产品经混合溶剂重结晶得到纯度98.5%以上的产品。
由于粗产品的纯度较高,重结晶母液中主要成分也是产品,所以可以用作水解工艺中第一次中和的中和剂。其原理为母液中的3-叔丁基-4-羟基苯丙酸可以中和酚羟基和过量的水解剂lioh,本身变为产品的锂盐而得到回收,提高产品总收率。
本发明的有益效果:
1、采用酯基的选择性水解方法及部分烷基化方法提高了烷基化反应的选择性,使产物的选择性达到95%以上。
2、采用两次酸化和有机溶剂萃取方法,简化了未反应原料及副产物与产物的分离工艺,提高了粗产品的质量。
3、采用混合溶剂重结晶技术,提高了产品质量。本发明工艺生产的产品为白色晶体,纯度达98.5%以上。
4、采用了未反应原料及重结晶母液回收技术,使产品总收率达到90%以上。
附图说明
图1是实施例一中步骤一脱烷基化反应高效液相色谱图。
图2是实施例一中步骤二萃取获得有机相高效液相色谱图。
图3是实施例一中步骤三中3-叔丁基-4-羟基苯丙酸粗品高效液相色谱图。
图4是实施例一中步骤三重结晶操作后得到的3-叔丁基-4-羟基苯丙酸高效液相色谱图。
图5是实施例一中步骤三重结晶母液的高效液相色谱图。
图6是实施例一中制备得到的3-叔丁基-4-羟基苯丙酸的核磁共振氢谱图。
图7是脱烷基反应中杂质2的核磁共振氢谱图。
具体实施方式
本发明技术方案不局限于以下所列举具体实施方式,还包括各具体实施方式间的任意组合。
具体实施方式一:本实施方式3-叔丁基-4-羟基苯丙酸的制备方法,包括以下步骤:
一、脱烷基化反应:将3,5-二叔丁基-4-羟基苯丙酸甲酯与催化剂加入反应器中,加热升温至60~90℃,hplc监控反应,当中间体质量百分含量达到65%~80%时停止反应;其中催化剂的质量为3,5-二叔丁基-4-羟基苯丙酸甲酯质量的1.5%~3%;所述中间体为3-叔丁基-4-羟基苯丙酸甲酯;
二、选择性水解:向步骤一所述的反应器中加入氢氧化锂水溶液,在60~80℃条件下反应至体系呈澄清状态,然后用稀盐酸调节水解体系ph为6.5~7.5,向体系中加入有机溶剂萃取未反应的原料,得到水相和有机相,将有机相回收溶剂得到有机相浓缩物;
三、分离提纯:将步骤二得到的水相用稀盐酸酸化至ph为1-2,析出白色固体,过滤,得3-叔丁基-4-羟基苯丙酸粗品;
将3-叔丁基-4-羟基苯丙酸粗品用乙腈-正丁醇混合液重结晶,得到的白色固体部分是纯品3-叔丁基-4-羟基苯丙酸,将重结晶的母液回收溶剂得到母液回收物备用;
四、回收利用:将步骤二得到的有机相浓缩物作为脱烷基化反应原料,回收反应,具体如下:
将步骤二得到的有机相浓缩物与催化剂加入反应器中,加热升温至60~90℃,hplc监控反应,当中间体质量百分含量达到65%~80%时停止反应;其中催化剂的质量为步骤二得到的有机相浓缩物质量的1.5%~3%;所述中间体为3-叔丁基-4-羟基苯丙酸甲酯;
然后向反应器中加入氢氧化锂水溶液,在60~80℃条件下反应至体系呈澄清状态,然后加入步骤三重结晶的母液回收物,再用稀盐酸调节水解体系ph为6.5~7.5,向体系中加入有机溶剂萃取,得到水相;
将水相用稀盐酸酸化至ph为1-2,析出白色固体,过滤,得3-叔丁基-4-羟基苯丙酸粗品;将3-叔丁基-4-羟基苯丙酸粗品用乙腈-正丁醇混合液重结晶,得到的白色固体部分是纯品3-叔丁基-4-羟基苯丙酸;
五、收集步骤三和步骤四得到的纯品3-叔丁基-4-羟基苯丙酸,即制备过程完成。
具体实施方式二:本实施方式与具体实施方式一不同的是:步骤一和步骤四中所述催化剂为路易斯酸,所述路易斯酸为对甲苯磺酸、甲基磺酸、磷酸或硫酸。其它与具体实施方式一相同。
具体实施方式三:本实施方式与具体实施方式一或二不同的是:步骤二和步骤四中所述氢氧化锂水溶液的质量百分浓度为10%-20%。其它与具体实施方式一或二相同。
具体实施方式四:本实施方式与具体实施方式一至三之一不同的是:步骤二中氢氧化锂与原料3,5-二叔丁基-4-羟基苯丙酸甲酯的摩尔比为(2.0-2.2)∶1。其它与具体实施方式一至三之一相同。
具体实施方式五:本实施方式与具体实施方式一至四之一不同的是:步骤二和步骤四中萃取所用的有机溶剂为乙酸乙酯、甲苯、正丁醇、石油醚、二氯甲烷或正己烷。其它与具体实施方式一至四之一相同。
具体实施方式六:本实施方式与具体实施方式一至五之一不同的是:步骤二中有机溶剂的体积与原料3,5-二叔丁基-4-羟基苯丙酸甲酯的质量比为(0.5~5.0)ml:1g。其它与具体实施方式一至五之一相同。
具体实施方式七:本实施方式与具体实施方式一至六之一不同的是:步骤三和步骤四所述乙腈-正丁醇混合液中乙腈与正丁醇的质量比为(0.6~1.0)∶1。其它与具体实施方式一至六之一相同。
具体实施方式八:本实施方式与具体实施方式一至七之一不同的是:步骤三中3-叔丁基-4-羟基苯丙酸粗品的质量与乙腈-正丁醇混合液的体积比为1g:(1.0-2.5)ml。其它与具体实施方式一至七之一相同。
具体实施方式九:本实施方式与具体实施方式一至八之一不同的是:步骤四中氢氧化锂与原料有机相浓缩物的摩尔比为(2.0-2.2)∶1。其它与具体实施方式一至八之一相同。
具体实施方式十:本实施方式与具体实施方式一至九之一不同的是:步骤四中有机溶剂的体积与原料有机相浓缩物的质量比为(0.5~5.0)ml:1g。其它与具体实施方式一至九之一相同。
具体实施方式十一:本实施方式与具体实施方式一至十之一不同的是:步骤四中3-叔丁基-4-羟基苯丙酸粗品的质量与乙腈-正丁醇混合液的体积比为1g:(1.0-2.5)ml。其它与具体实施方式一至十之一相同。
下面对本发明的实施例做详细说明,以下实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方案和具体的操作过程,但本发明的保护范围不限于下述的实施例。
实施例一:
本实施例为3-叔丁基-4-羟基苯丙酸的制备方法,具体的通过以下步骤实现的:
一、脱烷基化反应:将292g(1mol)3,5-二叔丁基-4-羟基苯丙酸甲酯与8.8g催化剂对甲苯磺酸加入反应器中,加热升温至90℃反应。hplc监控反应,当原料含量小于20%,中间体含量达到80%时结束反应。
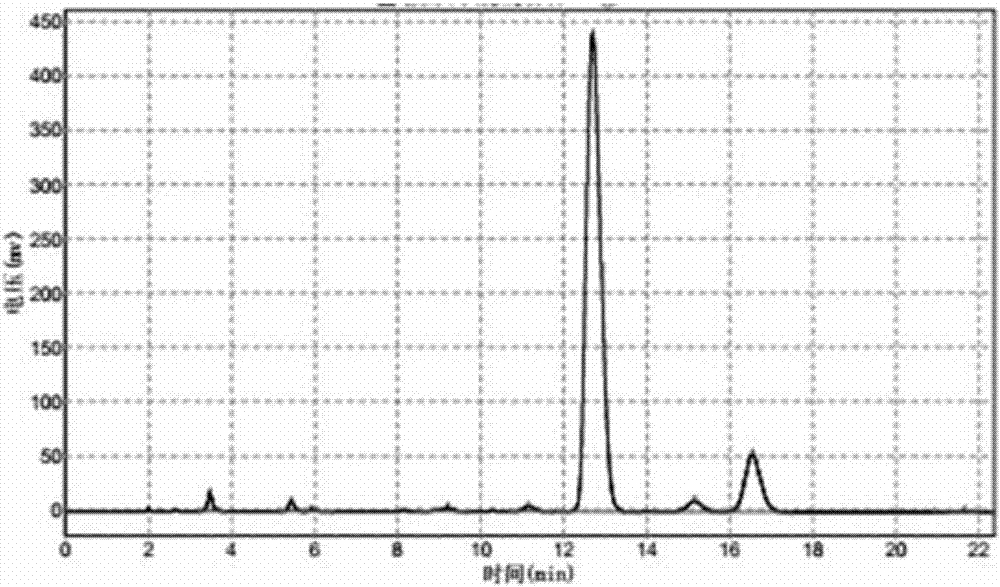
步骤一脱烷基化反应高效液相色谱图如图1所示。其中,杂质1:tr=3.208,含量4.14%;杂质3:tr=3.493,含量1.60%;中间体:tr=5.385,含量80.08%;原料:tr=12.470,含量10.96%;杂质2:tr=16.067,含量2.35%。
二、选择性水解:将52.0g氢氧化锂配成质量浓度为20%的氢氧化锂水溶液,将氢氧化锂水溶液加入到步骤一的反应器中,70℃下反应至体系变澄清。用稀盐酸调节水解体系ph为7,向体系中加入290ml正己烷萃取未反应的原料,得到的正己烷溶液回收溶剂作为有机相浓缩物。
步骤二萃取获得有机相高效液相色谱图如图2所示。其中,杂质3:tr=3.482,含量1.03%;原料:tr=12.698,含量83.92%;杂质2:tr=16.548,含量11.01%。
三、分离纯化:将步骤二中得到的水相用稀盐酸进行酸化至ph为2,析出白色固体,过滤,得3-叔丁基-4-羟基苯丙酸粗品195g,纯度96.8%。
步骤三中3-叔丁基-4-羟基苯丙酸粗品的高效液相色谱图如图3所示。其中,产品3-叔丁基-4-羟基苯丙酸:tr=3.480,含量96.80%。
将3-叔丁基-4-羟基苯丙酸粗品用210ml乙腈-正丁醇混合液重结晶,得白色固体3-叔丁基-4-羟基苯丙酸188g,纯度98.6%,步骤三重结晶操作后得到的3-叔丁基-4-羟基苯丙酸高效液相色谱图如图4所示,其中产品3-叔丁基-4-羟基苯丙酸:tr=3.505,含量98.59%。重结晶母液回收溶剂后作为母液回收物,步骤三重结晶母液的高效液相色谱图如图5所示,其中,产品3-叔丁基-4-羟基苯丙酸:tr=3.493,含量87.04%。其中乙腈-正丁醇混合液中乙腈和正丁醇的质量比为0.8∶1。
四、回收利用:将步骤二中有机相浓缩物加入催化剂后继续反应,重复步骤一~步骤三。其中,在步骤二中水解反应结束后,加入上一次重结晶母液回收物,再用稀盐酸调节体系ph为7。完成上述步骤后,共获得产品211g,总收率为95%。
本实施例制备得到的3-叔丁基-4-羟基苯丙酸的核磁共振氢谱图如图6所示,3-叔丁基-4-羟基苯丙酸的核磁氢谱数据分析如下:1hnmr(300mhz,cd3od,单位:ppm):7.05(d,j=2.2hz,1h),6.85(dd,j=2.3,8.1hz,1h),6.64(d,j=8.1hz,1h),2.81(t,j=7.5hz,2h),2.54(t,j=7.8hz,2h),1.38(s,9h)。由核磁氢谱数据确定,得到的产品为3-叔丁基-4-羟基苯丙酸。
另外,对脱烷基化反应中杂质2(即中间体之间的酯化产物)的结构进行确定,脱烷基反应中杂质2的核磁共振氢谱图如图7所示,核磁氢谱数据分析如下:1hnmr(300mhz,cdcl3,单位:ppm):7.21(d,j=2.0hz,1h),7.17(d,j=2.1hz,1h),7.04(dd,j=2.3,8.3hz,1h),6.98(dd,j=2.1,7.9hz,1h),6.82(d,j=8.1hz,1h),6.62(d,j=8.1hz,1h),5.10(s,1h),3.71(s,3h),3.04(t,j=7.3hz,2h),2.90(quint,j=7.3hz,4h),2.64(t,j=7.4hz,2h),1.43(s,9h),1.32(s,9h)。由此确定,杂质2是中间体之间的酯化产物。
本实施例中,采用高效液相色谱法对反应进程及产物纯度进行监控,条件如下:c18柱;检测波长uv=238nm;流动相:乙腈:水=75∶25,并加入流动相总体积1%的冰乙酸;流速v=1.0ml/min。保留时间:3,5-二叔丁基-4-羟基苯丙酸甲酯tr=12.470min,3-叔丁基-4-羟基苯丙酸甲酯tr=5.385min,3-叔丁基-4-羟基苯丙酸tr=3.480min。
- 还没有人留言评论。精彩留言会获得点赞!