一种左旋葡萄糖酮的制备方法与流程
本发明属于生物质能的利用技术领域,具体涉及一种高产率左旋葡萄糖酮的制备方法。
背景技术:
左旋葡萄糖酮(lgo,1,6-脱水-3,4-二脱氧-β-d-吡喃糖烯-2-酮)是一种纤维素热解形成的脱水糖产物,由于具有6,8-二氧二环[3.2.1]辛烷的烯酮体系、高活性的酮基以及缩醛中心,lgo可广泛用于多种合成反应,用于制备各种糖类和非糖类衍生物以及治疗癌症、免疫性疾病和心脏病等的药物。目前,还没有找到一种合适的方法规模化地生产lgo,导致其价格极为昂贵。
在纤维素热解中,若有酸性催化剂存在,可以得到一种高值化合物:左旋葡萄糖酮(levoglucosenone,lgo)。左旋葡萄糖酮是一种重要的手性合成子,是生物质资源开发与利用的又一新平台。内醚糖烯酮也可以采用化学法合成,但步骤比较繁杂,且原料昂贵,分离和纯化成本也较高,很难实现规模化生产。由纤维素在酸催化条件下进行热解通过控制条件可以得到左旋葡萄糖酮,产率与所用酸有很大关系。例如:在真空条件下磷酸作催化剂热解纤维素可以得到产率8%左右的lgo,利用硫酸催化产率则有所降低。蒙脱石作为路易斯酸优化条件下催化热解纤维素可以得到2.9%的lgo产率。研究表明,其他固体酸(al2o3、mgo、cro3、cuso4等)对lgo也没有太高的选择性,除了生成lgo外,还包括乙酸、糠醛、lg等其他复杂的组分。金属氯化物也作为催化剂被使用,但lgo的产率低于3%。利用so42-/zro2固体超强酸,可以获得8%左右的lgo产率,热解液的相对含量最高可达60%以上。用炭磺酸或磁性炭磺酸为催化剂,可以获得20%左右的lgo产率,但炭磺酸或磁性炭磺酸的制备需要大量的浓硫酸,造成浓硫酸的浪费和环境污染,不利于环保。
技术实现要素:
本发明要解决的技术问题是:根据现有左旋葡萄糖酮制备技术产率较低的不足,本发明提供一种高产率左旋葡萄糖酮的制备方法。本发明所选用的试剂为价廉的常规试剂,用量少;并且在反应过程中包括酸碱中和步骤,没有酸碱废物的排放,保护了环境,有利于环保,具有显著的经济效益和社会效益。
为了解决上述问题,本发明采取的技术方案是:
本发明提供一种左旋葡萄糖酮的制备方法,所述制备方法包括以下步骤:
a、首先将磷酸溶于水中形成磷酸溶液,然后在所得磷酸溶液中加入纤维素或含纤维素生物质,加入后进行充分混合,浸泡0.1~0.5h;
所述纤维素或含纤维素生物质与所得磷酸溶液之间加入量的固液质量比为1:5~20;
b、将步骤a浸泡后的纤维素或含纤维素生物质进行过滤、压挤或者离心,处理后将所得物质在100~280℃条件下进行热解预处理0.1~1h,得到脱水中间产物;
c、将所得脱水中间产物冷却到室温,冷却后置于氢氧化钠或氢氧化钾水溶液中充分混合,浸泡0.1~0.5h;
所述脱水中间产物与氢氧化钠或氢氧化钾水溶液之间加入量的固液质量比为1:5~20;
d、将步骤c中混合、浸泡后的产物进行过滤、压挤或离心,处理后将产物在300~400℃条件下进行无氧热解0.5~1h,热解过程中用<0℃的循环冷却液冷凝收集,得到含左旋葡萄糖酮的热解液。
根据上述的左旋葡萄糖酮的制备方法,步骤a中形成磷酸溶液的浓度为0.5~2.5mol/l。
根据上述的左旋葡萄糖酮的制备方法,步骤a中所述纤维素为脱脂棉或微晶纤维素;所述含纤维素生物质为甘蔗渣、秸秆或锯末。
根据上述的左旋葡萄糖酮的制备方法,步骤c中所述氢氧化钠或氢氧化钾水溶液的浓度为0.01~0.1mol/l。
根据上述的左旋葡萄糖酮的制备方法,步骤d中所述循环冷却液为冷却盐水或防冻液。
根据上述的左旋葡萄糖酮的制备方法,步骤d中所述无氧是指在惰性气氛或真空条件下。
本发明的积极有益效果:
1、本发明制备过程中,使用过的磷酸溶液经补充浓磷酸使之浓度保持在使用范围,可反复一直作为预处理浸泡液使用;使用后的氢氧化钠(或氢氧化钾)溶液经补充后使之浓度保持在使用范围,可反复作为中和浸泡液一直使用,酸碱溶液经上述步骤达到重复利用的目的,因此不存在酸碱排放问题,没有酸碱废物排放,有利于环保。
2、本发明在反应过程中包括酸碱中和步骤,所以没有酸碱废物的排放,有利于环保,具有显著的经济效益和社会效益。
3、本发明使用的原材料磷酸、氢氧化钠或氢氧化钾为常见化工原料,不需使用难以制备或不稳定的固体酸催化剂,因此原料价廉易得、操作简单。
4、本发明制备左旋葡萄糖酮采用两步反应,大大提高了产率(产率最高可达40%左右),相比现有技术成本大大降低。因此,本发明技术方案具有极大的应用前景。
附图说明:
图1本发明实施例1所得产品与标准品左旋葡萄糖酮的质谱图。
图1中:a为本发明实施例1所得产品左旋葡萄糖酮;b:标准品左旋葡萄糖酮。由图1本发明所得产品的质谱图与标准品左旋葡萄糖酮的质谱图相比,表明本发明所得产品为左旋葡萄糖酮。
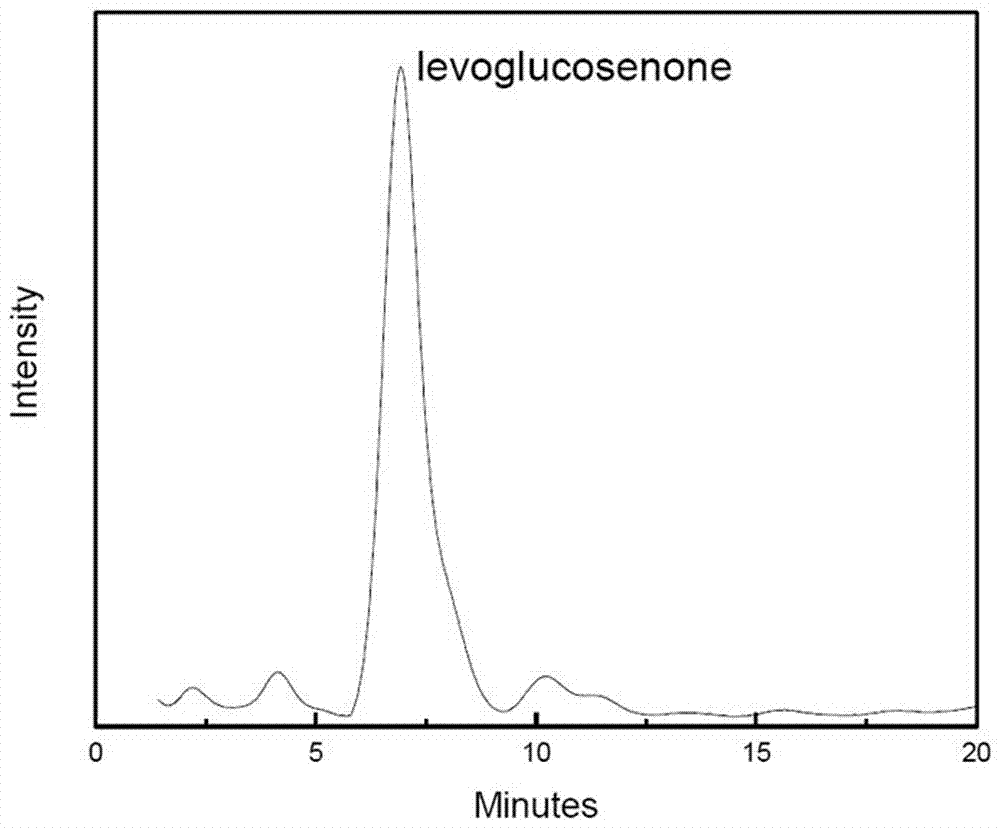
图2本发明实施例1所得产品的气相色谱图。
图2中所得产品为微晶纤维素热解液制备得到的左旋葡萄糖酮;从图2中可以看出本发明所得左旋葡萄糖酮的纯度较高。
具体实施例方式:
以下结合实施例进一步阐述本发明,但并不限制本发明保护的技术内容。
实施例1:
本发明左旋葡萄糖酮的制备方法,该制备方法的详细步骤如下:
a、首先将磷酸溶于水中形成2.0mol/l的磷酸溶液100ml,然后在所得磷酸溶液中加入10g微晶纤维素,加入后进行充分混合,浸泡10min;
b、将浸泡后的物质进行过滤(经过滤除去浸泡后微晶纤维素中吸附的大部分液体),然后将所得滤饼置于热解炉中,在真空(5mmhg)、200℃条件下进行热解预处理10min,得到脱水中间产物;
c、将所得脱水中间产物冷却到室温,冷却后置于100ml、0.05mol/l的氢氧化钠水溶液中充分混合,浸泡10min;
d、将步骤c所得产物进行过滤(经过滤除去所得产物中吸附的大部分液体),然后将所得滤饼置于热解炉中,在380℃条件下进行真空无氧热解30min(真空度为5mmhg),热解过程中用<0℃的循环冷却盐水冷凝收集,收集得到18.5ml冷凝液,经色谱分析左旋葡萄糖酮产率为41.5%。
本实施例使用过的磷酸溶液经补充浓磷酸使之浓度保持在2.0mol/l,可一直作为预处理浸泡液使用,使用后的氢氧化钠溶液经补充后使之浓度保持在0.05mol/l,可作为中和浸泡液一直使用,酸碱溶液经上述步骤达到重复利用的目的,因此不存在酸碱排放问题。
据报道,本发明现有技术中以“微晶纤维素”热解制备的左旋葡萄糖酮的最高产率为22.1%。
实施例2:
本发明左旋葡萄糖酮的制备方法,该制备方法的详细步骤如下:
a、首先将磷酸溶于水中形成1.5mol/l的磷酸溶液100ml,然后在所得磷酸溶液中加入10g微晶纤维素,加入后进行充分混合,浸泡20min;
b、将浸泡后的物质进行过滤(经过滤除去浸泡后微晶纤维素中吸附的大部分液体),然后将所得滤饼置于热解炉中,在真空(5mmhg)、150℃条件下进行热解预处理30min,得到脱水中间产物;
c、将所得脱水中间产物冷却到室温,冷却后置于100ml、0.03mol/l的氢氧化钠水溶液中充分混合,浸泡20min;
d、将步骤c所得产物进行过滤(经过滤除去所得产物中吸附的大部分液体),然后将所得滤饼置于热解炉中,在350℃条件下进行真空无氧热解40min(真空度为5mmhg),热解过程中用<0℃的循环冷却盐水冷凝收集,收集得到18.1ml冷凝液,经色谱分析左旋葡萄糖酮产率为40.8%。
本实施例使用过的磷酸溶液经补充浓磷酸使之浓度保持在1.5mol/l,可一直作为预处理浸泡液使用,使用后的氢氧化钠溶液经补充后使之浓度保持在0.03mol/l,可作为中和浸泡液一直使用,酸碱溶液经上述步骤达到重复利用的目的,因此不存在酸碱排放问题。
实施例3:
本发明左旋葡萄糖酮的制备方法,该制备方法的详细步骤如下:
a、首先将磷酸溶于水中形成2.2mol/l的磷酸溶液100ml,然后在所得磷酸溶液中加入10g微晶纤维素,加入后进行充分混合,浸泡10min;
b、将浸泡后的物质进行过滤(经过滤除去浸泡后微晶纤维素中吸附的大部分液体),然后将所得滤饼置于热解炉中,在真空(真空度为5mmhg)、220℃条件下进行热解预处理10min,得到脱水中间产物;
c、将所得脱水中间产物冷却到室温,冷却后置于100ml、0.08mol/l的氢氧化钠水溶液中充分混合,浸泡10min;
d、将步骤c所得产物进行过滤(经过滤除去所得产物中吸附的大部分液体),然后将所得滤饼置于热解炉中,在400℃条件下进行真空无氧热解30min(真空度为5mmhg),热解过程中用<0℃的循环冷冻液冷凝收集,收集得到19.0ml冷凝液,经色谱分析左旋葡萄糖酮产率为42.3%。
本实施例使用过的磷酸溶液经补充浓磷酸使之浓度保持在2.2mol/l,可一直作为预处理浸泡液使用,使用后的氢氧化钠溶液经补充后使之浓度保持在0.08mol/l,可作为中和浸泡液一直使用,酸碱溶液经上述步骤达到重复利用的目的,因此不存在酸碱排放问题。
实施例4:
本发明左旋葡萄糖酮的制备方法,该制备方法的详细步骤如下:
a、首先将磷酸溶于水中形成1.0mol/l的磷酸溶液100ml,然后在所得磷酸溶液中加入10g脱脂棉,加入后进行充分混合,浸泡20min;
b、将浸泡后的物质进行过滤(经过滤除去浸泡后脱脂棉中吸附的大部分液体),然后将所得滤饼置于热解炉中,在真空(真空度为5mmhg)、150℃条件下进行热解预处理20min,得到脱水中间产物;
c、将所得脱水中间产物冷却到室温,冷却后置于100ml、0.01mol/l的氢氧化钾水溶液中充分混合,浸泡20min;
d、将步骤c所得产物进行过滤(经过滤除去所得产物中吸附的大部分液体),然后将所得滤饼置于热解炉中,在360℃条件下进行真空无氧热解40min(真空度为5mmhg),热解过程中用<0℃的循环冷却盐水冷凝收集,收集得到16.5ml冷凝液,经色谱分析左旋葡萄糖酮产率为36.5%。
本实施例使用过的磷酸溶液经补充浓磷酸使之浓度保持在1.0mol/l,可一直作为预处理浸泡液使用,使用后的氢氧化钠溶液经补充后使之浓度保持在0.01mol/l,可作为中和浸泡液一直使用,酸碱溶液经上述步骤达到重复利用的目的,因此不存在酸碱排放问题。
实施例5:
本发明左旋葡萄糖酮的制备方法,该制备方法的详细步骤如下:
a、首先将磷酸溶于水中形成2.0mol/l的磷酸溶液100ml,然后在所得磷酸溶液中加入10g预先水洗干燥后的甘蔗渣,加入后进行充分混合,浸泡20min;
b、将浸泡后的物质进行过滤(经过滤除去浸泡后甘蔗渣中吸附的大部分液体),然后将所得滤饼置于热解炉中,在真空(真空度为5mmhg)、200℃条件下进行热解预处理10min,得到脱水中间产物;
c、将所得脱水中间产物冷却到室温,冷却后置于100ml、0.1mol/l的氢氧化钠水溶液中充分混合,浸泡20min;
d、将步骤c所得产物进行过滤(经过滤除去所得产物中吸附的大部分液体),然后将所得滤饼置于热解炉中,在380℃条件下进行真空无氧热解30min(真空度为5mmhg),热解过程中用<0℃的循环冷却盐水冷凝收集,收集得到12.0ml冷凝液,经色谱分析左旋葡萄糖酮产率为22.8%。
本实施例使用过的磷酸溶液经补充浓磷酸使之浓度保持在2.0mol/l,可一直作为预处理浸泡液使用,使用后的氢氧化钠溶液经补充后使之浓度保持在0.1mol/l,可作为中和浸泡液一直使用,酸碱溶液经上述步骤达到重复利用的目的,因此不存在酸碱排放问题。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种高效扁柏双黄酮的制备方法与流程
- (1R,4S)‑(-)‑2‑氮杂双环[2.2.1]庚‑5‑烯‑3‑酮的制备方法与制造工艺
- 制备环状和非环状酮的方法与制造工艺
- 7‑溴‑5‑(2‑氯苯基)‑1,3‑二氢噻吩并[2,3e]‑1,4‑二氮杂卓‑2‑硫酮的制备方法与制造工艺
- 一种2‑苯基‑1,4‑苯并哌喃酮的制备方法与制造工艺
- 一种2′4′‑二氟‑2‑[1‑(1H‑1,2,4‑三唑基)]苯乙酮的制备方法与制造工艺
- 一种5‑氨基苯并咪唑酮的制备方法与制造工艺
- 一种制备GLYT-1抑制剂的方法与制造工艺
- 一种1‑[5‑(苯基甲氧基)‑2‑吡啶]‑乙酮的制备方法与制造工艺
- 一种2‑氯乙酰氨基‑5硝基二苯甲酮的制备方法与制造工艺