一种利格列汀新晶型及其制备方法与流程
本发明涉及药物化学
技术领域:
,具体涉及一种利格列汀新晶型及其制备方法。
背景技术:
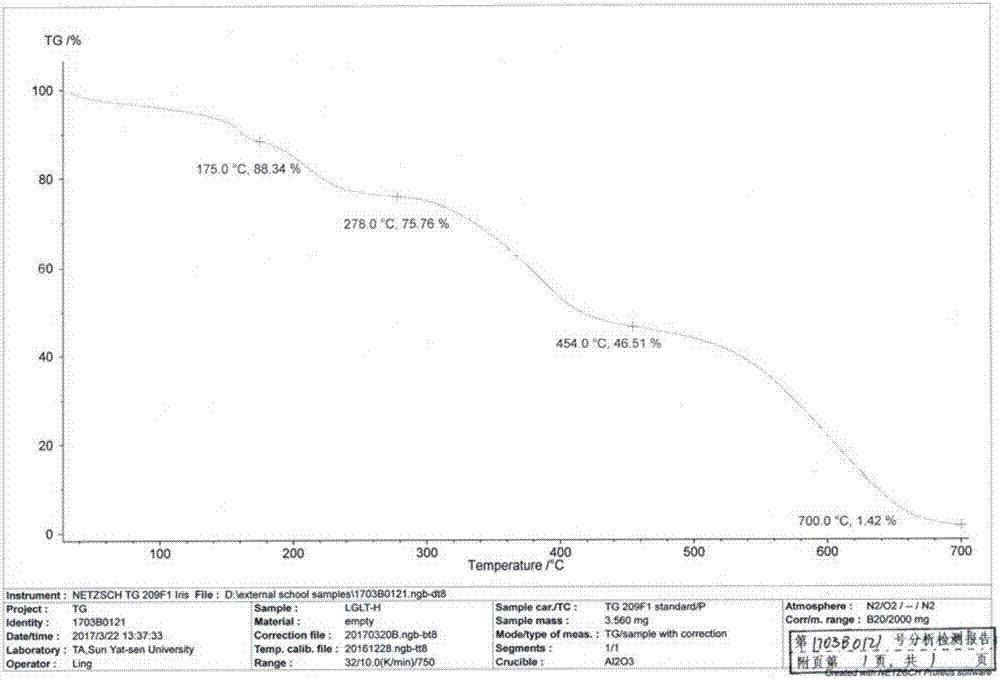
:利格列汀(linagliptin),由勃林格殷格翰研发的一种抑制二肽基肽酶-4抑制剂,用于治疗2型糖尿病,于2011年5月获得fda批准。利格列汀化学名为8-(3r)-氨基-哌啶-1-基-7-丁-2-炔-3-甲基-1-((4-甲基喹唑啉-2-基)-甲基)-黄嘌呤,其结构式如式ⅰ所示:据现有专利文献报道利格列汀具有无定型和其他结晶晶型,但是药用使用的是结晶形式的利格列汀。其中(1)us2007/0259900a1公开了利格列汀a、b、c、d、e五种晶型及其制备方法,并且该公司使用稳定形态a和c晶型为其药用晶型,其中,晶型b由晶型a在<10℃的情况下转化的;晶型d是由晶型c加热到30℃-100℃转化而来,存在晶型转化的问题,晶型e则是由晶型d熔融得到的,不能适应规模化生产。(2)cn105272982a公开了一种晶型,通过溶剂溶解再用反溶剂析晶而制备。(3)cn104418857a公开的是利格列汀的无定型态。(4)wo2013/128379a2公开了两种晶型,通过乙醇/水和乙醇/醚得到。(5)us2013/0123282a1公开了通过各种溶剂试验得到了28种利格列汀的晶型。技术实现要素:为了克服现有技术的不足,本发明的目的在于提供一种利格列汀新晶型及其制备方法,该晶型定义为晶型h,该新晶型具有良好的化学稳定性和纯度,易于规模化制备,操作简单,具有广阔的应用前景。为解决上述问题,本发明所采用的技术方案如下:一种利格列汀的新晶型h,其如式i所示:1)其差热分析显示在0~180℃之间有两个吸收峰,分别在46±3℃和162±3℃,其中162±3℃吸收峰为主要吸收峰;2)热重分析显示不含有结晶水或溶剂化合物;3)熔点为150~153℃。优选地,本发明所述的新晶型h,使用cu-kα射线,以2θ±0.2角度表示x射线衍射在4.0°,8.0°,9.5°,11.9°,16.0°,18.8°,19.9°,21.5°,22.5°,24°处有衍射峰。更优选地,本发明所述的新晶型h,使用cu-kα射线,以2θ±0.2角度表示x射线衍射在4.0°,8.0°,9.5°,11.5°,11.9°,12.4°,16.0°,16.9°,18.3°,18.8°,19.9°,21.5°,22.5°,24°处有衍射峰。在本发明优选的实施方案中,新晶型h具有如图1所示的xrd图谱。本发明所述的新晶型h的x-射线粉末衍射图具有如下特征:序号2-thetaheightfwhmd-spacingrel.int%area14.0511047.940.102321.8121100.00105.7928.011230.570.204711.036522.0046.5539.513241.000.15359.297523.0036.49411.512125.970.15357.686812.0219.07511.976198.950.20477.390318.9940.17612.456121.800.15357.106611.6218.44714.9350.820.15355.93264.857.70816.026185.530.17915.530317.7032.78916.46498.730.15355.38449.4214.951016.905124.650.17915.244911.8922.021118.340148.760.17914.837614.2026.281218.842162.520.15354.709715.5124.611319.540114.490.15354.543210.9317.341419.930121.830.15354.455911.6318.451521.571296.660.15354.119628.3144.921622.529229.010.12793.946721.8528.901724.060191.080.15353.698918.2328.931824.99788.830.20473.56238.4817.931926.36847.700.40933.38024.5519.262028.70854.420.15353.10975.198.242130.68749.880.30702.91354.7615.10本发明所述的利格列汀新晶型h,通过熔点仪测量,熔程为150~153℃。熔程是指物质的初熔温度到终熔温度之间的温度区间,初熔温度即物质开始熔解的温度,终熔温度即物质完全熔解的温度。一般地,熔程越短,纯度越高。本发明所述的新晶型h经红外吸收分析,具有如下特征吸收峰:1698,1655,1613,1570,1508,1435,765。在本发明优选的实施方案中,利格列汀新晶型h具有如图2所示的红外吸收光谱。本发明所述的利格列汀新晶型h,经差示扫描量热法测定在30~50℃及150~170℃有吸热峰,其吸热峰的顶值在46±3℃和162±3℃。在本发明优选的实施案例中,利格列汀新晶型h的差示扫描量热图如图3所示。本发明所述的利格列汀新晶型h,加热至175℃有约12%的质量损失,随后在278℃、454℃直到700℃基本分解完毕,由此判断该晶型不含结晶水或结晶溶剂。在本发明的具体实施案例中,利格列汀新晶型h的热重分析图如图4所示。本发明所述的利格列汀新晶型h,在具体的实施案例中,利格列汀晶型h的核磁共振氢谱(1h-nmr)如图6所示、核磁共振碳谱(13c-nmr)如图7所示。本发明还提供了如上所述的利格列汀新晶型h的制备方法,其包括以下步骤:(1)将8-(3r)-氨基-哌啶-1-基-7-丁-2-炔-3-甲基-1-((4-甲基喹唑啉-2-基)-甲基)-黄嘌呤加入二氯甲烷中,溶清,浓缩至干得到无定型粗品;(2)向无定型粗品中加入溶剂i,加热溶解,室温下搅拌析晶,过滤,干燥,得到利格列汀新晶型h。优选地,步骤(2)中所述溶剂i为二甲基亚砜与二氧六环的混合溶剂,二甲基亚砜与二氧六环的混合比例为5:1~1:5,优选1:1;溶剂i与无定型粗品的质量体积比为2:1~10:1,优选4:1,单位为ml/g。相比现有技术,本发明的有益效果在于:经由本发明所述的制备方法制得的利格列汀新晶型h经x-射线粉末衍射、红外吸收分析、差热分析、热重分析、核磁共振及质谱确证,其中热重分析表明新晶型h不含水和溶剂,差热分析表明所得晶型h为单一晶型,x-射线粉末衍射表明与文献报道的晶型及无定型完全不同,为一种新的晶型。该晶型具有良好的化学稳定性和晶型纯度,易于规模化制备,操作简单,具有广阔的应用前景。附图说明图1为本发明所述的利格列汀晶型h的xrd图谱;图2为本发明所述的利格列汀晶型h的红外吸收光谱图谱;图3为本发明所述的利格列汀晶型h的dsc图谱;图4为本发明所述的利格列汀晶型h的tga图谱;图5为本发明所述的利格列汀晶型h的显微镜图;图6为本发明所述的利格列汀晶型h的1hnmr图谱;图7为本发明所述的利格列汀晶型h的13cnmr图谱;图8为利格列汀晶型a的xrd图谱;图9为利格列汀晶型c的xrd图谱。具体实施方式下面结合附图和具体实施方式对本发明作进一步详细说明。需要说明的是,在本发明中,如未作特殊说明,溶剂与相关试剂的用量为反应的常规用量,本领域的技术人员根据现有技术即可确定;本发明使用的试剂为常规试剂,可通过市场购买得到,所用起始原料和反应物均可通过现有技术或公开的现有文献制备得到。除特殊说明外,本发明所述“室温”具有本领域的公知含义,具体是指10~30℃,优选20~30℃。无定型粗品的制备:将20g8-(3r)-氨基-哌啶-1-基-7-丁-2-炔-3-甲基-1-((4-甲基喹唑啉-2-基)-甲基)-黄嘌呤固体溶于100ml二氯甲烷中,充分搅拌溶解后,浓缩至干,得利格列汀的无定形态20g,即无定型粗品。实施例1:利格列汀晶型h的制备取已制备的无定型粗品5g,加10ml二甲基亚砜,加热至50℃溶解,取出冷却至室温,缓慢搅拌下加入10ml二氧六环,待有固体析出后,降温0~5℃,继续搅拌1h。过滤,45℃真空干燥,得类白色晶体3.2g。收率为64%,hplc纯度为98.78%,水分0.3%。经测定,其xrd图谱与图1基本一致;其红外光谱与图2基本一致;通过熔点仪测量其熔程为:150℃~153℃;其dsc图谱与图3基本一致;其tga图谱与图4基本一致;在显微镜下观察,晶型h如图5所示。实施例2:利格列汀晶型h的制备取已制备的无定型粗品2g,加10ml二甲基亚砜,加热至40℃溶解,取出冷却至室温,缓慢搅拌下加入5ml二氧六环,待有固体析出后,降温0~5℃,继续搅拌1h。过滤,45℃真空干燥,得类白色晶体1g。收率50%,hplc纯度为98.99%,水分0.6%。经测定,其xrd图谱与图1基本一致;其红外光谱与图2基本一致;通过熔点仪测量其熔程为:150℃~153℃;其dsc图谱与图3基本一致;其tga图谱与图4基本一致;在显微镜下观察,晶型h如图5所示。实施例3:利格列汀晶型h的制备取已制备的无定型粗品3g,加20ml二甲基亚砜,加热至40℃溶解,取出冷却至室温,缓慢搅拌下加入2ml二氧六环,待有固体析出后,降温0~5℃,继续搅拌3h。过滤,50℃真空干燥,得类白色晶体1.36g。收率为45.3%,hplc纯度为99.2%,水分为0.5%。经测定,其xrd图谱与图1基本一致;其红外光谱与图2基本一致;通过熔点仪测量其熔程为:150℃~153℃;其dsc图谱与图3基本一致;其tga图谱与图4基本一致;在显微镜下观察,晶型h如图5所示。实施例4:利格列汀晶型h的制备取已制备的无定型粗品5g,加15ml二甲基亚砜,加热至45℃溶解,取出冷却至室温,缓慢搅拌下加入5ml二氧六环,待有固体析出后,继续搅拌2h。过滤,50℃真空干燥,得类白色晶体3.12g。收率为62.4%,hplc纯度为98.71%,水分为0.7%。经测定,其xrd图谱与图1基本一致;其红外光谱与图2基本一致;通过熔点仪测量其熔程为:150℃~153℃;其dsc图谱与图3基本一致;其tga图谱与图4基本一致;在显微镜下观察,晶型h如图5所示。实施例5:利格列汀晶型h的制备取已制备的无定型粗品3g,加6ml二甲基亚砜,加热至60℃溶解,取出冷却至室温,缓慢搅拌下加入12ml二氧六环,待有固体析出后,降温0~5℃,继续搅拌2h。过滤,40℃真空干燥,得类白色晶体1.68g。收率56.1%,hplc纯度98.75%,水分0.4%。经测定,其xrd图谱与图1基本一致;其红外光谱与图2基本一致;通过熔点仪测量其熔程为:150℃~153℃;其dsc图谱与图3基本一致;其tga图谱与图4基本一致;在显微镜下观察,晶型h如图5所示。实施例6:利格列汀晶型h的制备取已制备的无定型粗品5g,加10ml二甲基亚砜,加热至50℃溶解,取出冷却至室温,缓慢搅拌下加入5ml二氧六环,待有固体析出后,继续搅拌1h。过滤,40℃真空干燥,得类白色晶体2.82g。收率56.4%,hplc纯度为98.72%,水分0.7%。经测定,其xrd图谱与图1基本一致;其红外光谱与图2基本一致;通过熔点仪测量其熔程为:150℃~153℃;其dsc图谱与图3基本一致;其tga图谱与图4基本一致;在显微镜下观察,晶型h如图5所示。实施例7:利格列汀晶型a的制备取已制备的无定型粗品5g,加25ml乙醇,加热至50℃溶解,取出冷却至室温,缓慢搅拌下加入50ml叔丁基甲基醚,析出固体后,降温0~5℃,继续搅拌1h。过滤,45℃真空干燥,得类白色晶体4.1g。经测定,其xrd图谱与图8基本一致。实施例8::利格列汀晶型c的制备取已制备的无定型粗品5g,加12.5ml甲醇,加热至50℃溶解,取出冷却至室温,缓慢搅拌下加入50ml叔丁基甲基醚,析出固体后,降温0~5℃,继续搅拌1h。过滤,45℃真空干燥,得类白色晶体3.8g。经测定,其xrd图谱与图9基本一致。由利格列汀新晶型h的制备实施例1~6与利格列汀晶型a实施例7、利格列汀晶型c实施例8的xrd图谱对比可知,本发明所制得的晶型h与目前文献报道的稳定晶型及无定型完全不同,为一种新的晶型。利格列汀新晶型h稳定性试验:取实施例1制备得到的利格列汀新晶型h样品放置在35℃的条件下,考察在放置1个月、2个月、3个月的稳定性,试验结果见表1。具体的稳定性考察的方法可以参照中国药典2010版第二部附录xixc的方法;纯度检测用hplc法,可以参照中国药典2010版第二部附录vd的方法。表1利拉利汀新晶型h的稳定性试验结果由上述实验可以看出,本发明制备得到的利格列汀新晶型h经过3个月稳定性试验,样品的纯度、水分、晶型均未发生改变,说明本发明提供的利格列汀新晶型h稳定,且具有良好的化学稳定性。上述实施方式仅为本发明的优选实施方式,不能以此来限定本发明保护的范围,本领域的技术人员在本发明的基础上所做的任何非实质性的变化及替换均属于本发明所要求保护的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1