一种聚氨基酸嵌段共聚物及其制备方法和应用与流程
本发明属于高分子材料领域,尤其涉及一种聚氨基酸嵌段共聚物及其制备方法和应用。
背景技术:
原位给药抗肿瘤系统是指将药物或药物载体直接注射到肿瘤部位,从而提高肿瘤部位药物浓度,降低药物的全身分布。对比于传统的静脉全身给药,原位给药系统在病灶部分持续的释放出药物,直接作用于肿瘤部位,避免了全身给药药物在非肿瘤部位的聚集,从而提高药物传输效果,降低药物对其他器官的毒副作用。原位给药系统具有用药剂量少、用药次数少、疗效高、药物对正常细胞组织的毒副作用小等特点,成为近20年来药学领域的热门研究课题。然而,原位给游离药常常不能达到理想效果,因为游离药扩散很快,不易在肿瘤部位长时间聚集。通过可植入物担载化疗药物进行原位治疗,又由于其对机体损伤较大,成本较高,而不能被广泛应用。因此,如何实现方便快捷的原位可持续的给药,成为原位给药系统的一大难题。
温度敏感型水凝胶具有独特的溶液-凝胶转变特性,控制它的相变温度与人体体温匹配,即可得到温度敏感可注射型水凝胶:在低于人体体温时,温度敏感可注射型水凝胶以溶液状态存在,可以与药物、多肽、蛋白质、细胞等药物或生物活性物质混合;当注入到肿瘤附近后,由于温度的变化,溶液迅速发生相变,形成水凝胶,在形成水凝胶的过程中,混合在其中的药物或生物活性物质被包埋在水凝胶内部,然后通过扩散或水凝胶自身的降解缓慢释放,从而达到在肿瘤部位长效缓释的目的。这类水凝胶具有流动性好,使用方便,滞留时间长,释药缓慢、持久的特点,而且它对低分子溶质具有较好的透过性,有优良的生物相容性和较好的重现性,容易合成,因此近年来被广泛用于生物医学领域,尤其在蛋白质和多肽类药物的缓控释给药系统中的应用引起了众多药学研究者的关注。
聚合物可形成温度敏感型水凝胶,成为了近年来的研究热点之一。其中,聚氧化乙烯-聚氧化丙烯缩合物(也称poloxamer,泊洛沙姆)的水溶液可用作温度敏感可注射型水凝胶,它是目前研究最多的一类用作温度敏感型水凝胶的聚合物,但是它在体内只能存在数天即被体液稀释,无法实现长期持续给药,而且泊洛沙姆不能生物降解,这就限制了其在人体内的应用;随后,发展了一种聚乙二醇和聚(l-乳酸)嵌段共聚物的水凝胶,它可以被生物降解,但是其降解产物为乳酸等小分子化合物,局部过高浓度的乳酸会引起炎症,对人体的健康不利。
技术实现要素:
有鉴于此,本发明的目的在于提供一种聚氨基酸嵌段共聚物及其制备方法和应用,采用本发明提供的聚氨基酸嵌段共聚物制成的温敏性水凝胶具有良好的生物降解性,且降解产物对人体无不利影响。
本发明提供了一种聚氨基酸嵌段共聚物,具有式(i)或式(ii)结构:
其中,m、n和j为聚合度,10≤m≤200,0≤n≤41,2≤j≤101。
优选的,50≤m≤150;2≤n≤8;5≤j≤50。
本发明提供了一种聚氨基酸嵌段共聚物的制备方法,包括以下步骤:
端氨基聚合物、l-苯丙氨酸-n-羧酸内酸酐和l-丙氨酸-n-羧酸内酸酐进行聚合反应,得到具有式(i)或式(ii)结构的聚氨基酸嵌段共聚物;所述端氨基聚合物具有式(iii)或式(iv)结构;
其中,m、n和j为聚合度,10≤m≤200,0≤n≤41,2≤j≤101。
优选的,所述聚合反应的温度为20~40℃;所述聚合反应的时间为24~72h。
本发明提供了一种温敏性水凝胶,包括聚氨基酸嵌段共聚物和溶剂;所述聚氨基酸嵌段共聚物为上述技术方案所述的聚氨基酸嵌段共聚物或上述技术方案所述方法制备的聚氨基酸嵌段共聚物。
优选的,所述溶剂包括水、生理盐水、缓冲溶液、组织培养液或体液。
优选的,所述聚氨基酸嵌段共聚物在温敏性水凝胶中的含量为2~30wt%。
本发明提供了一种原位抗肿瘤组合物,包括抗肿瘤药物和上述技术方案所述的温敏性水凝胶。
优选的,所述抗肿瘤药物包括阿霉素、表阿霉素、吡喃阿霉素、康普他汀、康普他汀磷酸二钠、甲氨蝶呤、紫杉醇、多西紫杉醇、顺铂、卡铂、奥沙利铂、硼替佐米、喜树碱和紫草素中的一种或多种。
优选的,所述抗肿瘤药物在组合物中的含量为2~30wt%。
与现有技术相比,本发明提供了一种聚氨基酸嵌段共聚物及其制备方法和应用。本发明提供的聚氨基酸嵌段共聚物具有式(i)或式(ii)结构,由该共聚物和溶剂混合制成的水凝胶具有温敏性,在较低温度时,其为水溶液;当温度升高至人体温度附近时,水溶液即可转变成凝胶。而且由该共聚物和溶剂混合制成的水凝胶具有良好的生物降解性,其降解周期约为2周~15周,降解产物为氨基酸和聚乙二醇,可通过肾脏直接排出体外,对人体无不利影响。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据提供的附图获得其他的附图。
图1是本发明实施例15提供的聚氨基酸嵌段共聚物的核磁共振氢谱图;
图2是本发明实施例35提供的温敏性聚氨基酸水凝胶的的成胶相图;
图3是本发明实施例35提供的温敏性聚氨基酸水凝胶的生物降解曲线;
图4是本发明实施例38提供的原位抗肿瘤组合物溶液-固体转变拍摄图;
图5是本发明实施例52提供的原位抗肿瘤组合物的药物释放图。
具体实施方式
下面对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明提供了一种聚氨基酸嵌段共聚物,具有式(i)或式(ii)结构:
其中,m、n和j为聚合度,10≤m≤200,0≤n≤41,2≤j≤101。
本发明提供的聚氨基酸嵌段共聚物具有式(i)或式(ii)结构,其中m、n和j为聚合度。在本发明中,10≤m≤200,优选为50≤m≤150,更优选为100≤m≤120,m具体可选择为12、24、48、96或192。在本发明中,0≤n≤41,优选为0≤n≤10,更优选为2≤n≤8,最优选为2≤n≤6,n具体可选择为0、1.7、1.9、2、2.1、2.3、2.7、4、4.3、8.1、8.2、8.4、20.3或40.9。在本发明中,2≤j≤101,优选为5≤j≤50,更优选为5≤j≤20,j具体可选择为2.2、2.5、4.8、4.9、5、5.1、5.2、5.4、5.5、5.7、10.2、10.3、10.9、11.2、12.1、12.4、12.5、12.6、12.7、12.8、20.3、20.5、21.5、24.2、24.9、44.8、47.1、95.7或100.7。
本发明提供的聚氨基酸嵌段共聚物具有式(i)或式(ii)结构,由该共聚物和溶剂混合制成的水凝胶具有温敏性,在较低温度时,其为水溶液;当温度升高至人体温度附近时,水溶液即可转变成凝胶。而且由该共聚物和溶剂混合制成的水凝胶具有良好的生物降解性,其降解周期约为2周~15周,降解产物为氨基酸和聚乙二醇,可通过肾脏直接排出体外,对人体无不利影响。
本发明提供了一种聚氨基酸嵌段共聚物的制备方法,包括以下步骤:
端氨基聚合物、l-苯丙氨酸-n-羧酸内酸酐和l-丙氨酸-n-羧酸内酸酐进行聚合反应,得到具有式(i)或式(ii)结构的聚氨基酸嵌段共聚物;所述端氨基聚合物具有式(iii)或式(iv)结构;
其中,m、n和j为聚合度,10≤m≤200,0≤n≤41,2≤j≤101。
在本发明提供的制备方法中,直接将端氨基聚合物、l-苯丙氨酸-n-羧酸内酸酐和l-丙氨酸-n-羧酸内酸酐进行聚合反应,即可得到具有式(i)或式(ii)结构的聚氨基酸嵌段共聚物。具体可以按照以下方式进行制备:
首先,提供第一溶液和第二溶液,第一溶液包括端氨基聚合物和第一溶剂,第二溶液包括l-苯丙氨酸-n-羧酸内酸酐、l-丙氨酸-n-羧酸内酸酐和第二溶剂。在本发明中,所述端氨基聚合物具有式(iii)或式(iv)结构。其中,式(iii)结构端氨基聚合物为端氨基化的聚乙二醇,其结构如下:
其中,m为聚合度,10≤m≤200。
在本发明中,式(iv)结构端氨基聚合物为端氨基化的聚乙二醇单甲醚,其结构如下:
其中,m为聚合度,10≤m≤200。
在本发明中,所述第一溶剂包括但不限于n,n-二甲基甲酰胺、n,n-二甲基乙酰胺和三氯甲烷中的一种或多种。本发明对所述端氨基聚合物和第一溶剂的用量比没有特别限定,可选择为(0.1~10)g:100ml,具体可选择为0.5g:100ml、1g:100ml、5g:100ml或10g:100ml。
在本发明中,端氨基聚合物在溶解于第一溶剂之前,先进行前处理。所述前处理的具体过程包括:先将端氨基聚合物与无水甲苯混合,共沸除水;之后除去甲苯。其中,所述端氨基聚合物与无水甲苯用量比优选为(0.1~10)g:200ml,具体可选择为0.5g:200ml、1g:200ml、5g:200ml或10g:200ml;所述共沸除水的温度优选为110-150℃,更优选为115-140℃,最优选为125-135℃,具体可为130℃;所述共沸除水的时间优选为1-3h,更优选为1.5-2.5h,最优选为1.8-2.2h,具体可为2h。
在本发明中,l-苯丙氨酸-n-羧酸内酸酐的结构如下:
在本发明中,l-丙氨酸-n-羧酸内酸酐的结构如下:
在本发明中,所述第二溶剂包括但不限于n,n-二甲基甲酰胺、n,n-二甲基乙酰胺和三氯甲烷中的一种或多种。本发明对所述l-苯丙氨酸-n-羧酸内酸酐、l-丙氨酸-n-羧酸内酸酐和第二溶剂的用量比没有特别限定,所述l-苯丙氨酸-n-羧酸内酸酐、l-丙氨酸-n-羧酸内酸酐和第二溶剂的用量比可选择为(0~5)g:(0.1~26)g:100ml,具体可选择为0.87g:0g:100ml、1.73g:0g:100ml、0g:1.04g:100ml、0g:0.52g:100ml、0g:0.26g:100ml、0g:0.13g:100ml、0g:0.65g:100ml、0g:5.4g:100ml、0g:2.6g:100ml、0g:3.2g:100ml、0.87g:3.2g:100ml、1.73g:3.2g:100ml、3.46g:3.2g:100ml、0.87g:6.4g:100ml、0.87g:13.8g:100ml、0.87g:13g:100ml、0.87g:0.05g:100ml、1.73g:0.32g:100ml或0.87g:26g:100ml。
准备好第一溶液和第二溶液后,将第一溶液和第二溶液混合。其中,混合得到的混合体系中端氨基聚合物、l-苯丙氨酸-n-羧酸内酸酐和l-丙氨酸-n-羧酸内酸酐的质量比优选为(0.1~10):(0~5):(0.1~26),具体可选择为1:0:1.04、1:0:0.52、1:0:0.26、1:0:0.13、1:0.87:0、1:1.73:0、1:0.87:0.05、1:1.73:0.32、10:0:0.65、10:0:5.4、10:0:2.6、10:0:0.32、10:0.87:3.2、10:1.73:3.2、10:3.46:3.2、10:0.87:6.4、10:0.87:13、10:0.87:26、0.5:0:1.04、0.5:0:0.52、0.5:0:0.26、0.5:0:0.13、5:0:0.65、5:0:5.4、5:0:2.6、5:0:0.65、5:0:3.2、5:0.87:0.32、5:1.73:0.32、5:3.46:3.2、5:0.87:6.4、5:0.87:13.8、或5:0.87:26。第一溶液和第二溶液混合后,混合体系中端氨基聚合物、l-苯丙氨酸-n-羧酸内酸酐和l-丙氨酸-n-羧酸内酸酐进行聚合反应。其中,所述聚合反应的温度优选为20~40℃;所述聚合反应的时间优选为24~72h;所述聚合反应优选在氮气保护条件下进行。聚合反应结束后,得到的反应液进行后处理,得到聚氨基酸嵌段共聚物。在本发明提供的一个实施例中,所述后处理的过程具体包括:首先除去反应液中的溶剂,得到固体;然后用氯仿溶解所述固体,得到溶液;之后用乙醚对其进行沉降,得到沉降物;接的对沉降物依次进行抽滤、洗涤和干燥,得到聚氨基酸嵌段共聚物。
采用本发明提供的方法能够制备得到式(i)或式(ii)结构的聚氨基酸嵌段共聚物,由该共聚物和溶剂混合制成的水凝胶具有温敏性,在较低温度时,其为水溶液;当温度升高至人体温度附近时,水溶液即可转变成凝胶。而且由该共聚物和溶剂混合制成的水凝胶具有良好的生物降解性,其降解周期约为2周~15周,降解产物为氨基酸和聚乙二醇,可通过肾脏直接排出体外,对人体无不利影响。
本发明提供的一种温敏性水凝胶,包括聚氨基酸嵌段共聚物和溶剂;所述聚氨基酸嵌段共聚物为上述技术方案所述的聚氨基酸嵌段共聚物或上述技术方案所述方法制备的聚氨基酸嵌段共聚物。
本发明提供的温敏性水凝胶包括所述聚氨基酸嵌段共聚物和溶剂。其中,溶剂包括但不限于水、生理盐水、缓冲溶液、组织培养液或体液,优选为缓冲溶液。所述缓冲溶液优选包括磷酸盐缓冲溶液或tris-hcl缓冲溶液。在本发明中,所述聚氨基酸嵌段共聚物在温敏性水凝胶中的含量优选为2~30wt%,更优选为4~20wt%,最优选为5~15%,具体可选择为2wt%、3wt%、4wt%、5wt%、6wt%、7wt%、8wt%、9wt%、10wt%、11wt%、12wt%、13wt%、14wt%、15wt%、16wt%、17wt%、18wt%、19wt%、20wt%、21wt%、22wt%、23wt%、24wt%、25wt%、26wt%、27wt%、28wt%、29wt%或30wt%。
本发明对所述温敏性水凝胶的制备方法没有特别限定,直接将包括所述聚氨基酸嵌段共聚物和溶剂的原料混合均匀即可。其中,所述混合的温度优选为2~10℃,更优选为4~6℃;所述混合的方式优选为搅拌;所述搅拌的时间优选为5~48h,更优选为10~36h,,最优选为24h。
本发明提供的水凝胶具有温敏性,在较低温度时,其为水溶液;当温度升高至人体温度附近时,水溶液即可转变成凝胶。而且该水凝胶具有良好的生物降解性,其降解周期约为2周~15周,降解产物为氨基酸和聚乙二醇,可通过肾脏直接排出体外,对人体无不利影响。
本发明提供了一种原位抗肿瘤组合物,包括抗肿瘤药物和上述技术方案所述的温敏性水凝胶。
本发明提供的原位抗肿瘤组合物包括抗肿瘤药物和所述温敏性水凝胶。其中,所述抗肿瘤药物包括但不限于阿霉素、表阿霉素、吡喃阿霉素、康普他汀、康普他汀磷酸二钠、甲氨蝶呤、紫杉醇、多西紫杉醇、顺铂、卡铂、奥沙利铂、硼替佐米、喜树碱和紫草素中的一种或多种。在本发明中,所述抗肿瘤药物在组合物中的含量为优选为2~30wt%,更优选为4~20wt%,最优选为5~15%,具体可选择为2wt%、3wt%、4wt%、5wt%、6wt%、7wt%、8wt%、9wt%、10wt%、11wt%、12wt%、13wt%、14wt%、15wt%、16wt%、17wt%、18wt%、19wt%、20wt%、21wt%、22wt%、23wt%、24wt%、25wt%、26wt%、27wt%、28wt%、29wt%或30wt%。
本发明对所述原位抗肿瘤组合物的制备方法没有特别限定,直接将包括抗肿瘤药物、溶剂和所述聚氨基酸嵌段共聚物的原料混合均匀即可。其中,所述混合的温度优选为2~10℃,更优选为4~6℃;所述混合的方式优选为搅拌;所述搅拌的时间优选为5~48h,更优选为10~36h,,最优选为24h。
本发明提供的原位抗肿瘤组合物具有温敏性,在低于人体体温时,以溶液状态存在;当注入到肿瘤附近后,由于温度的变化,溶液迅速发生相变,形成凝胶。在形成凝胶的过程中,混合在其中的抗肿瘤药物被包埋在凝胶内部,然后通过扩散或凝胶自身的降解缓慢释放,从而达到在肿瘤部位长效缓释的目的。本发明提供的原位抗肿瘤组合物在降解过程中产生的降解产物为氨基酸和聚乙二醇,可通过肾脏直接排出体外,对人体无不利影响。
为更清楚起见,下面通过以下实施例进行详细说明。
下述实施例中,所述端氨基化的聚乙二醇具有式(iii)结构;所述端氨基化的聚乙二醇单甲醚具有式(iv)结构;缓冲溶液为磷酸盐缓冲溶液,其中磷酸二氢钾浓度为0.24gl-1,磷酸二氢钠浓度为1.42gl-1,氯化钠浓度为8.0gl-1,氯化钾浓度为0.2gl-1。
实施例1
向干燥的反应瓶内加入1g、数均分子量为550(即聚合度为12)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将1.04gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=12、n=0、j=4.9。
实施例2
向干燥的反应瓶内加入1g、数均分子量为1100(即聚合度为24)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.52gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=24、n=0、j=5。
实施例3
向干燥的反应瓶内加入1g、数均分子量为2200(即聚合度为48)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.26gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=48、n=0、j=5.1。
实施例4
向干燥的反应瓶内加入1g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.13gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=0、j=5。
实施例5
向干燥的反应瓶内加入10g、数均分子量为8800(即聚合度为192)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.65gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=192、n=0、j=4.8。
实施例6
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将5.4gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=0、j=20.3。
实施例7
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将2.6gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=0、j=10.2。
实施例8
向干燥的反应瓶内加入1g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.05gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=20.3、j=2.2。
实施例9
向干燥的反应瓶内加入1g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.32gl-丙氨酸-n-羧酸内酸酐与1.73gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=40.9、j=12.1。
实施例10
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=2.1、j=12.6。
实施例11
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐与1.73gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=4、j=12.4。
实施例12
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐与3.46gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=8.2、j=11.2。
实施例13
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将6.4gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=2.3、j=24.2。
实施例14
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将13gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=2、j=48.8。
实施例15
向干燥的反应瓶内加入10g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将26gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,核磁共振氢谱检测结果如图1所示,图1是本发明实施例15提供的聚氨基酸嵌段共聚物的核磁共振氢谱图。核磁共振氢谱检测和聚合度测定结果显示该聚合物具有式(ii)结构,其中,m=96、n=1.7、j=100.7。
实施例16
向干燥的反应瓶内加入0.5g、数均分子量为550(即聚合度为12)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将1.04gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=12、n=0、j=5.5。
实施例17
向干燥的反应瓶内加入0.5g、数均分子量为1100(即聚合度为24)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.52gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=24、n=0、j=5.7。
实施例18
向干燥的反应瓶内加入0.5g、数均分子量为2200(即聚合度为48)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.26gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=48、n=0、j=5.5。
实施例19
向干燥的反应瓶内加入0.5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇单甲醚,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.13gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(ii)结构,其中,m=96、n=0、j=5.2。
实施例20
向干燥的反应瓶内加入5g、数均分子量为8800(即聚合度为192)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.65gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=192、n=0、j=5.4。
实施例21
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将5.4gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=0、j=21.5。
实施例22
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将2.6gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=8.1、j=10.3。
实施例23
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将0.65gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=0、j=2.5。
实施例24
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=0、j=12.8。
实施例25
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=2.1、j=12.7。
实施例26
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐与1.73gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=4.3、j=12.5。
实施例27
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将3.2gl-丙氨酸-n-羧酸内酸酐与3.46gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=8.4、j=10.9。
实施例28
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将6.4gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=2.7、j=24.9。
实施例29
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将13.8gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=1.9、j=47.1。
实施例30
向干燥的反应瓶内加入5g、数均分子量为4400(即聚合度为96)的端氨基化的聚乙二醇,与200ml无水甲苯在130℃下共沸除水2h后,减压抽干剩余的甲苯;将得到的固体溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第一溶液;将26gl-丙氨酸-n-羧酸内酸酐与0.87gl-苯丙氨酸-n-羧基内酸酐溶解于100ml干燥的n,n-二甲基甲酰胺中,得到第二溶液;在氮气氛围中,将第一溶液与第二溶液混合,在氮气保护条件下搅拌,40℃反应24h;反应结束后,减压抽干n,n-二甲基甲酰胺,然后将得到的固体溶解于氯仿中,再用乙醚进行沉降,抽滤,干燥后,得到聚氨基酸嵌段共聚物。
对上述聚氨基酸嵌段共聚物进行核磁共振氢谱检测和聚合度测定,结果显示该聚合物具有式(i)结构,其中,m=96、n=2.1、j=95.7。
实施例31
称取20mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入980mg水,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为51℃。白色固体。
实施例32
称取20mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入980mg生理盐水,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为56℃。白色固体。
实施例33
称取20mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入980mg缓冲溶液,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为52℃。白色固体。
实施例34
称取40mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入960mg缓冲溶液,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为33℃。白色固体。
实施例35
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入920mg缓冲溶液,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为22℃。白色固体。
1、采用小瓶倒置法确定聚氨基酸溶液的成胶温度,具体实验步骤如下:
1)配置不同浓度的聚氨基酸溶液,4℃下搅拌24。然后将得到的溶液转移到内径为11mm的小瓶中。
2)将装有不同浓度聚氨基酸溶液的小瓶放入恒温水浴中,调节所需温度,在设定温度下平衡5分钟。本实验取的浓度范围为4~20%质量分数。本实验设定温度范围为10~70℃。温度变化率为1℃每次。
3)取出小瓶,倒置小瓶,观察溶液有无流动,若溶液无流动,则该点温度即为成胶温度。
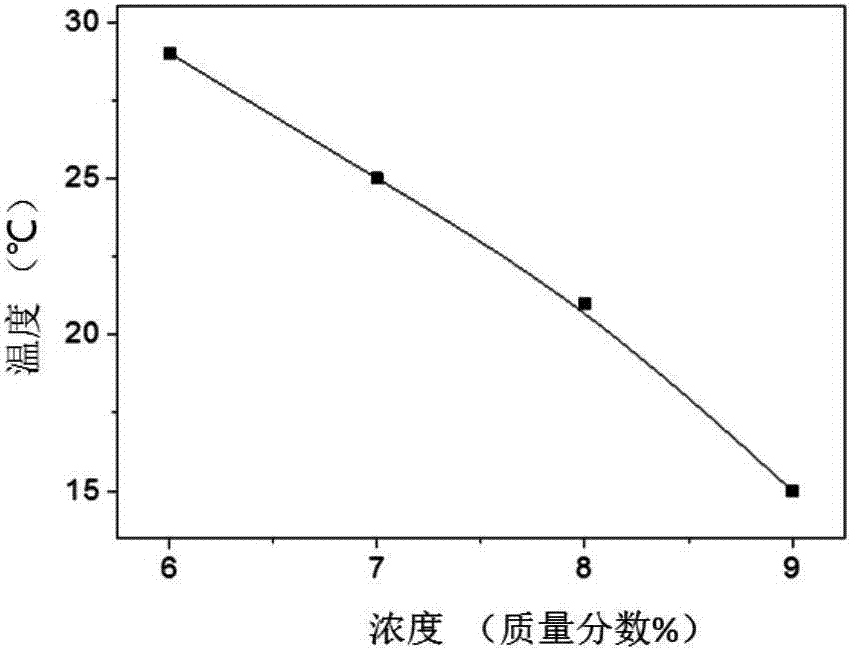
成胶相图如图2所示,图2是本发明实施例35提供的温敏性聚氨基酸水凝胶的的成胶相图。通过图2可以看出。实施例15提供的聚氨基酸嵌段共聚物在浓度为6~9wt%均可成胶,并且随着浓度增加,其成胶温度降低。
2、采用体外模拟生理环境的方法,确定聚氨基酸水凝胶的降解。具体步骤如下:
1)制备两组同样的聚氨基酸溶液,4℃搅拌24h。
2)将其放置在37℃恒温震荡箱中,震荡频率为70rpm,称重。
3)在这两组水凝胶中分别加入1ml磷酸盐缓冲溶液或含有5u弹性蛋白酶的磷酸盐缓冲溶液,置于37℃恒温震荡箱中,震荡频率70rpm。
4)每天将水凝胶取出,用滤纸吸去水凝胶表面溶液,称重。分别补加1ml的磷酸盐缓冲溶液或含有5u弹性蛋白酶的磷酸盐缓冲溶液。
5)计算每天损失的重量,根据时间作图,得到凝胶在在有或者没有弹性蛋白酶存在下的降解曲线。
生物降解曲线如图3所示,图3是本发明实施例35提供的温敏性聚氨基酸水凝胶的生物降解曲线。通过图3可以看出本发明提供的凝胶在模拟体内环境的加弹性蛋白酶的降解实验中,表现出很好的可降解性,进一步证明了我们的凝胶具有很好的生物可降解性。
实施例36
称取160mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入840mg缓冲溶液,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为15℃。白色固体。
实施例37
称取300mg实施例15中所述的聚氨基酸嵌段共聚物,置于干燥的小瓶中,在加入700mg缓冲溶液,放置于4℃,搅拌24h。及得到温敏性聚氨基酸水凝胶。成胶温度为10℃。白色固体。
实施例38
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为29℃。红色固体。
分别拍摄上述原位抗肿瘤组合物在4℃和37℃的照片,结果如图4所示,图4是本发明实施例38提供的原位抗肿瘤组合物溶液-固体转变拍摄图。通过图4可以看出,本发明提供的原位抗肿瘤组合物在4℃时为水溶液,37℃时转变成凝胶。
实施例39
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg康普他汀,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为30℃。白色固体。
实施例40
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg甲氨蝶呤,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为32℃。黄色固体。
实施例41
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg喜树碱,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为31℃。白色固体。
实施例42
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg紫杉醇,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为32℃。白色固体。
实施例43
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg顺铂,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为32℃。白色固体。
实施例44
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg奥沙利铂,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为34℃。白色固体。
实施例45
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg紫草素,置于干燥的小瓶中,在加入900mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为35℃。紫色固体。
实施例46
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取40mg阿霉素,置于干燥的小瓶中,在加入880mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为30℃。红色固体。
实施例47
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取80mg阿霉素,置于干燥的小瓶中,在加入840mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为24℃。红色固体。
实施例48
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取160mg阿霉素,置于干燥的小瓶中,在加入760mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为20℃。红色固体。
实施例49
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取300mg阿霉素,置于干燥的小瓶中,在加入620mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为16℃。红色固体。
实施例50
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,置于干燥的小瓶中,在加入880mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为34℃。红色固体。
实施例51
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,40mg康普他汀,置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为30℃。红色固体。
实施例52
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,80mg康普他汀,置于干燥的小瓶中,在加入820mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为26℃。红色固体。
共载药物的释放曲线通过体外模拟体内环境检测,具体实验方法如下:
1)制备两组同样的共载考布他汀与阿霉素的聚氨基酸溶液,4℃搅拌24h。
2)将其放置在37℃恒温震荡箱中,震荡频率为70rpm。
3)在这两组水凝胶中分别加入1ml磷酸盐缓冲溶液或含有5u弹性蛋白酶的磷酸盐缓冲溶液,置于37℃恒温震荡箱中,震荡频率70rpm。
4)每天将水凝胶取出,收集凝胶上缓冲溶液。再分别补加1ml的磷酸盐缓冲溶液或含有5u弹性蛋白酶的磷酸盐缓冲溶液。
5)通过标准曲线的方法,采用紫外-可见分光光度仪测试药物浓度。
6)通过药物浓度与取出液体含量,计算得到每天的药物释放量,根据时间作图得到图5,图5是本发明实施例52提供的原位抗肿瘤组合物的药物释放图。
通过图5可以看出,在酶的存在下,阿霉素的释放速率大于考布他汀,通过共载药物的方法,可以实现药物之间的释放速率的调控。
实施例53
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,160mg康普他汀,置于干燥的小瓶中,在加入740mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为20℃。红色固体。
实施例54
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,300mg康普他汀,置于干燥的小瓶中,在加入600mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为17℃。红色固体。
实施例55
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,20mg紫杉醇置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为36℃。红色固体。
实施例56
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,40mg紫杉醇置于干燥的小瓶中,在加入840mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为32℃。红色固体。
实施例57
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,80mg紫杉醇置于干燥的小瓶中,在加入800mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为30℃。红色固体。
实施例58
称取80mg实施例15中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,160mg紫杉醇置于干燥的小瓶中,在加入720mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为27℃。红色固体。
实施例59
称取80mg实施例1中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,300mg紫杉醇置于干燥的小瓶中,在加入580mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为53℃。红色固体。
实施例60
称取80mg实施例4中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,20mg紫杉醇置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为43℃。红色固体。
实施例61
称取80mg实施例8中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,20mg紫杉醇置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为36℃。红色固体。
实施例62
称取80mg实施例20中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,20mg紫杉醇置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为43℃。红色固体。
实施例63
称取80mg实施例25中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,20mg紫杉醇置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为27℃。红色固体。
实施例64
称取80mg实施例30中所述的聚氨基酸嵌段共聚物,称取20mg阿霉素,20mg康普他汀,20mg紫杉醇置于干燥的小瓶中,在加入860mg缓冲溶液,放置于4℃,搅拌24h。及得到原位抗肿瘤组合物。成胶温度为15℃。红色固体。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!