蝗虫微孢子虫实时荧光定量PCR检测方法及试剂盒与流程
本发明属于生物技术领域,涉及一种蝗虫微孢子虫实时荧光定量pcr检测方法及试剂盒,特别涉及一种蝗虫微孢子虫实时荧光定量pcrtaqman-mgb探针检测方法及试剂盒与应用。
背景技术:
自二十世纪八十年代以来,蝗虫微孢子虫(paranosemalocustae,p.l.)就被用来作为生物治蝗的重要生物制剂之一,世界各地都有使用且效果良好的报道。蝗虫感染微孢子虫的早期阶段没有明显的临床症状,喷洒微孢子制剂后,定期对蝗虫进行微孢子虫感染率检测,分析和掌握其扩散传播范围以及流行动态等十分必要。
目前,微孢子虫检测常用的方法有显微镜观察检测法和pcr方法。在家蚕等其它昆虫微孢子虫检测方法还有免疫学检测方法,但由于微孢子表面具有多抗原决定簇,假阳性比例较高,导致免疫学检测技术难以推广应用。而传统的显微镜检测技术,操作繁琐,灵敏度差,容易带来主观误差。普通pcr方法虽然快捷灵敏度高,但是容易交叉污染出现假阳性结果,且无法对样品进行定量。
荧光定量pcr技术是近年来发展起来的新型的核酸检测技术,该技术在普通pcr扩增时除了加入一对引物外还同时另外加入一个特异性的荧光探针,该探针只与靶基因特异性地结合,其结合位点在两条引物之间。荧光信号的强度就代表了模板dna的拷贝数,样本dna初始浓度的对数值与临界循环数(ct)值之间存在线性关系,通过比较扩增反应的ct值,分析反应模板的初始浓度,可达到精确定量扩增产物的目的。
技术实现要素:
本发明的目的是提供一种蝗虫微孢子虫实时荧光定量pcr检测方法及试剂盒。
首先,本发明提供了用于检测蝗虫微孢子虫的组合物。
本发明所提供的用于检测蝗虫微孢子虫的组合物,具体由一个引物对和一个探针组成;所述引物对由引物1和引物2组成,所述引物1为序列表中序列1所示的单链dna,所述引物2为序列表中序列2所示的单链dna;所述探针为序列表中序列3所示的单链dna探针。
其中,所述引物对和所述探针也分别属于本发明的保护范围。
所述探针的5’端标记有荧光基团,3’端标记有淬灭基团。
具体的,在本发明中,所述荧光基团为fam;所述淬灭基团为mgb。
其次,本发明提供了用于检测蝗虫微孢子虫的试剂盒。
本发明所提供的用于检测蝗虫微孢子虫的试剂盒,含有下述a)和b):
a)蝗虫微孢子虫质粒标准品;
b)前文所述的组合物或引物或探针。
其中,所述蝗虫微孢子虫质粒标准品为含有序列表中序列4所示dna片段的质粒。
具体的,所述蝗虫微孢子虫质粒标准品为将序列表中序列4所示dna片段与pmd18-t载体相连后得到的重组质粒。
在本发明的所述组合物中,所述引物1、所述引物2和所述探针的摩尔比具体可为1:1:1。
如下c)或d)的应用也属于本发明的保护范围:
c)前文所述的组合物或引物或探针在制备所述试剂盒中的应用;
d)前文所述的组合物或引物或探针或试剂盒在检测或辅助检测蝗虫微孢子虫中的应用。
在d)所述应用中,所述检测或辅助检测蝗虫微孢子虫为用前文所述的组合物或引物或探针或试剂盒对待测样品进行taqman探针荧光定量rt-pcr反应;
进行所述taqman探针荧光定量rt-pcr反应时,反应体系中所述引物1和所述引物2的浓度为200nm,所述探针的浓度为200nm。更加具体的,所述反应体系(20μl)如下:10μlfaststartessentialdnaprobesmaster,0.4μl上、下游引物(10μmol/l),0.4μl荧光标记探针(10μmol/l),1μldna模板,去离子水补齐。
进行所述taqman探针荧光定量rt-pcr反应时,扩增程序中的退火温度为60℃。更加具体的,所述扩增程序为:95℃2min;95℃10s,60℃60s,45个循环。
本发明以genbank数据库中公布的保守性高的微孢子虫小核糖体亚单位rna为目的基因,设计特异引物和taqman-mgb探针,通过制备重组质粒pmd18-t-p.l.作为标准品构建标准曲线,优化了引物和探针浓度及各反应参数,同时检测了该方法的灵敏性和重复性,首次建立了一种准确、方便和快捷的蝗虫微孢子虫taqman实时荧光定量pcr检测方法,为大规模样本的检测以及科学研究提供技术支持。
附图说明
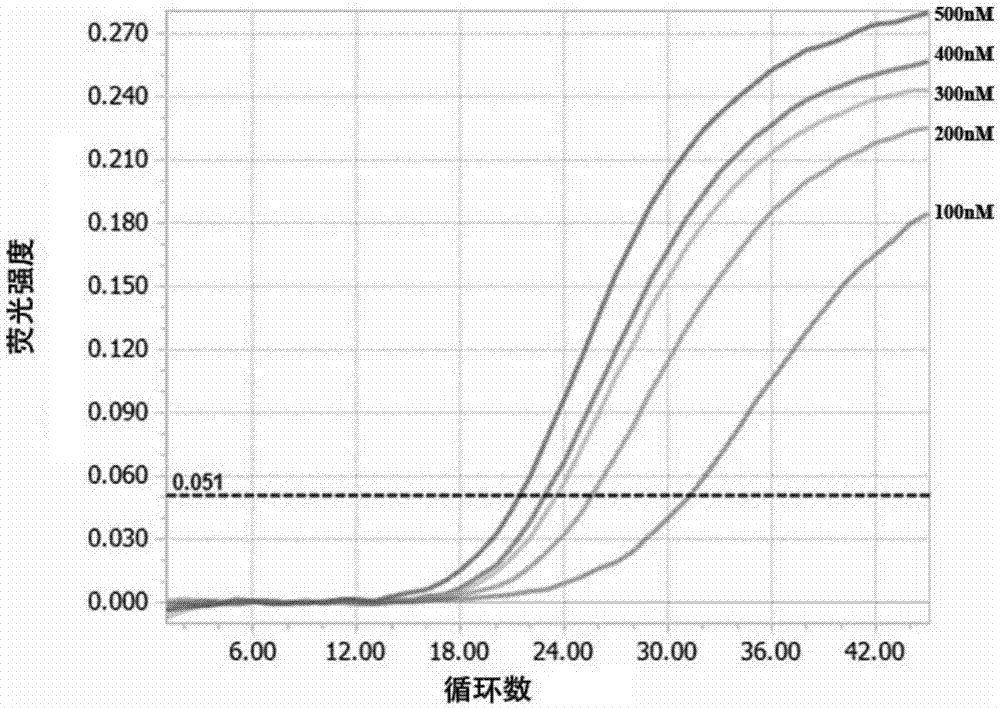
图1为不同探针浓度扩增曲线。
图2为不同引物浓度扩增曲线。
图3为荧光定量pcr的扩增曲线和标准曲线。
图4为不同稀释度标准质粒检测pcr扩增的敏感性。a:不同稀释度标准质粒荧光定量pcr的扩增曲线;b:不同稀释度标准质粒常规pcr电泳图。
图5为不同稀释度微孢子虫的常规pcr结果。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1、蝗虫微孢子虫实时荧光定量pcr检测方法的建立及应用
一、材料与方法
1.1微孢子虫
蝗虫微孢子虫(paranosemalocustae,p.l.)悬液(1×109p.l./ml)由中国农业大学农业部生物防治重点开放实验室提供。
1.2主要试剂及仪器
2×faststartessentialdnaprobesmaster为roche公司(美国)产品;dna凝胶回收试剂盒和质粒小量提取试剂盒为生工生物工程(上海)有限公司产品。taq酶、pmd18-t载体、dnamarkerdl2000均为takara公司产品。
lightcycler96荧光定量pcr仪为roche公司(美国)产品;nanodrop2000微量紫外分光度计为thermoscientific公司(美国)产品。
1.3引物与探针的设计
参照genbank数据库小孢子虫属的ssurrna序列,分析并设计引物和探针(表1),预计扩增的片段长度为117bp(序列4);taqman-p.l.探针5'端荧光基团为fam,3'端荧光淬灭基团为mgb,由上海生工生物技术有限公司合成。
表1荧光定量pcr扩增引物
1.4质粒标准品的制备
根据何永强等人[何永强,吴姗,鲁兴萌,邱海洪,帅江冰,张晓峰,王素华,徐国群,李光才,董强.不同dna抽提方法对普通pcr和实时荧光定量pcr方法检测家蚕微孢子虫的影响.昆虫学报,2011,54(11):1329-1334.]使用的方法提取微孢子虫的dna,经pcr方法扩增后使用割胶回收试剂盒进行凝胶回收,回收产物克隆至pmd18-t载体上,转化如大肠杆菌体内后,送含有阳性重组质粒的菌液测序。提取测序正确的重组质粒,用nd2000测定浓度,并根据拷贝数计算公式:[6.02×1023×(ng/l×10-9)]/(dna长度×660),计算每微升质粒含有的拷贝数。将该阳性质粒作为p.l.标准质粒。
1.5反应条件的优化
1.5.1探针浓度
固定引物浓度为500nmol/l,探针浓度分别为100、200、250、300、350、和400nm,每一个浓度平行做3孔,根据扩增反应的循环数(cyclethresholdvalue,ct值)和扩增曲线的荧光强度选择最优探针工作浓度。
1.5.2引物浓度
固定探针浓度为最优浓度,引物浓度为100、200、300、400和500nmol/l,每一个浓度平行做3孔,根据扩增反应的ct值和扩增曲线的荧光强度选择最优引物工作浓度。
1.6标准曲线的建立
将p.l.标准质粒进行10×倍比稀释,分别取1.29×1010-1.29×106拷贝/μl共5个浓度梯度的质粒为模板进行定量pcr,按照上述优化的体系进行反应,同时做3孔平行样。
1.7灵敏性检测
分别取1.29×1010-1.29×101拷贝/μl共10个浓度梯度的p.l.标准质粒为模板进行定量pcr和普通pcr,确定两种方法的检测下线并分析其敏感性。
1.8重复性检测
探针和引物分别以优化后的浓度进行荧光定量pcr反应体系,以p.l.标准质粒(1.29×1010-1.29×106拷贝/μl)为模板进行重复性检测。在同一次pcr反应中,每个稀释度标准质粒做6个重复孔,分析组内差异;用上述相同条件分别进行6次独立重复实验,分析组间差异,计算变异系数(coefficientofvariation,cv),cv=标准偏差(standarddeviation,sd)/平均数(mean)。
1.9反应体系及参数
定量pcr反应体系(20μl):10μlfaststartessentialdnaprobesmaster,0.4μl上、下游引物(10μmol/l),0.4μl荧光标记探针(10μmol/l),1μldna模板,去离子水补齐。扩增程序:95℃2min;95℃10s,60℃60s,45个循环。
常规pcr反应体系(20μl):2μlbuffer,0.4μl上、下游引物(10μmol/l),0.4μldntp,0.3μltaq酶,1μl模板,去离子水补齐。扩增程序:95℃3min;95℃30s,58℃30s,72℃10s,30个循环;72℃5min。
注:常规pcr所用的引物为上述表1中的上下游引物。
1.10实际样品的检测
取浓度为1×108孢子/ml微孢子虫原液进行2×倍比稀释,提取微孢子虫dna,分别利用上述建立的探针荧光定量pcr方法和普通pcr方法进行检测,并分析试验结果。
二、实验结果
2.1质粒标准品的制备
将微孢子虫浓缩液提取的总dna用普通引物(表1)进行pcr扩增,可得到117bp的扩增产物。将扩增产物与pmd18-t载体连接后得到重组质粒pmd18-t-p.l.。测序后序列与已知序列一致,证明成功构建了标准阳性模板重组质粒。根据拷贝数计算公式,得到该质粒浓度为1.29×1010拷贝/μl。
重组质粒pmd18-t-p.l.的结构描述为:将序列表中序列4所示dna片段与pmd18-t载体相连后得到的重组质粒。
2.2反应条件优化
2.1.1探针浓度优化
当探针浓度为100nmol/l时,ct值最大25.13,荧光强度最小;当探针浓度为350-400nmol/l时,ct值分别是15.86和17.67,虽然荧光强度较大,但是扩增曲线不稳定;当探针浓度为200-300nmol/l时,ct值在19.98-22.48之间,重复实验结果稳定。因此选择200nmol/l为探针的工作浓度(图1)。
2.1.2引物浓度优化
当引物浓度分别为100、200、300、400和500nmol/l时,其ct值在20.77-21.36之间。
当引物浓度为200nmol/l,可获得理想的扩增效果,扩增曲线呈现典型的“s”型(图2),因此选为引物工作浓度。
2.3标准曲线的绘制
取1.29×1010-1.29×106拷贝/μl共5个浓度梯度的质粒pmd18-t-p.l.作为模板进行real-timepcr,每个浓度重复3次进行检测,得到扩增曲线和标准曲线(图3)。起始模板浓度的对数值与ct值呈现线性关系,y=-3.1247x+33.541,扩增效率为108.93%,相关系数r2为0.9929。
2.4灵敏性检测
用10倍比稀释的标准质粒pmd18-t-p.l.(1.29×1010-1.29×101拷贝/μl)为模板,引物和探针浓度均为200nmol/l进行定量pcr。当模板浓度为1.29×105拷贝/μl时ct值为37.32,为最低检出浓度(图4中a),比常规pcr灵敏度高出100倍。在常规pcr扩增电泳图中,模板低于1.29×107拷贝/μl时没有可识别的目的片段(图4中b)。重复实验结果稳定不变。
2.5重复性检测
以1.29×1010-1.29×106拷贝/μl5个浓度的标准质粒pmd18-t-p.l.为模板进行重复性检测,结果如表2所示,各浓度标准质粒的组内和组间重复的变异系数均小于1%,表明该方法的重复性较好。
表2荧光定量pcr的重复性实验结果
2.6实际样品的检测
将浓度为1×108孢子/ml微孢子虫原液进行2×倍比稀释得到10个不同稀释度的微孢子虫液,分别提取dna,进行普通pcr检测,结果最低检出样品为8倍稀释的样品,有比较淡的大小为117bp的目的条带(图5);荧光定量pcr检测的最低检出样品为512倍稀释的样品,其ct为38.86(表3)并且有良好的扩增曲线。结果表明本发明建立的taqman荧光定量pcr检测方法比普通pcr方法灵敏度高64倍。
表3微孢子虫taqman荧光定量pcr检测结果
<110>新疆师范大学
<120>蝗虫微孢子虫实时荧光定量pcr检测方法及试剂盒
<130>gncln171036
<160>4
<170>patentinversion3.5
<210>1
<211>22
<212>dna
<213>人工序列
<220>
<223>
<400>1
gcctgacgtagacgctatactc22
<210>2
<211>27
<212>dna
<213>人工序列
<220>
<223>
<400>2
aacatagttataatcctaatacatcaa27
<210>3
<211>33
<212>dna
<213>人工序列
<220>
<223>
<400>3
tagacgctatactctaagattaacccatgcatg33
<210>4
<211>117
<212>dna
<213>人工序列
<220>
<223>
<400>4
gcctgacgtagacgctatactctaagattaacccatgcatgtttattgaatataaagaaa60
agacgaacagctcagtaactcttattttatttgatgtattaggattataactatgtt117
- 还没有人留言评论。精彩留言会获得点赞!
- 一种特异性引物检测鼠源性成分的方法与制造工艺
- 一组用于驴源性的实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一组用于鸡源性实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一种燕窝制品的实时荧光定量PCR鉴定试剂盒及其鉴定方法与制造工艺
- 一种具有移液功能的荧光定量PCR仪的制造方法与工艺
- 一种基于多灾种实时耦合的化工园区实时定量风险评估方法与制造工艺
- 一种实时荧光定量PCR检测方法及其标准品和检测试剂盒与制造工艺
- 检测外周血CAR‑T细胞的TaqMan实时荧光定量PCR试剂盒的制造方法与工艺
- 一种实时荧光定量PCR检测土壤西瓜枯萎病菌的方法及引物与制造工艺
- 一种基于反射镜的荧光定量PCR的检测系统的制造方法与工艺
- 一种基于荧光定量PCR鉴定啮齿类螺杆菌的引物、试剂盒及鉴定方法与流程
- 一种检测包含潜伏期结核感染的试剂盒及其引物对和探针的制造方法与工艺
- 用于甜瓜果实基因PCR表达分析的内参基因及该内参基因的稳定性验证方法与流程
- 一种特异性引物检测鼠源性成分的方法与制造工艺
- 一组用于驴源性的实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一组用于鸡源性实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一种燕窝制品的实时荧光定量PCR鉴定试剂盒及其鉴定方法与制造工艺
- 一种具有移液功能的荧光定量PCR仪的制造方法与工艺
- 一种基于多灾种实时耦合的化工园区实时定量风险评估方法与制造工艺
- 一种实时荧光定量PCR检测方法及其标准品和检测试剂盒与制造工艺
- TMEM209基因作为内参基因在定量检测稻螟赤眼蜂基因表达量中的应用的制作方法与工艺
- 蝗虫微孢子虫实时荧光定量PCR检测方法及试剂盒与流程
- ZFP基因作为内参基因在定量检测螟黄赤眼蜂基因表达量中的应用的制造方法与工艺
- 一种鸭新型腺病毒Eva Green实时荧光定量PCR检测的引物的制造方法与工艺
- 一种ampC基因的实时荧光定量PCR快速检测系统的制造方法与工艺
- RPS23基因作为内参基因在定量检测螟黄赤眼蜂基因表达量中的应用的制造方法与工艺
- 一种基于实时荧光PCR定量检测肉制品猪源性成分的方法与流程
- 一种用于基因测序的DNA荧光定量分析装置的制造方法
- 一种鲫鱼冠状病毒TaqMan实时荧光定量RT‑PCR检测引物及应用的制造方法与工艺
- 梨果实不同发育时期荧光定量内参基因的筛选及应用的制造方法与工艺