氨基正己酰氨基甲环酰氨基正己酰碱性氨基酸,其合成,活性和应用的制作方法
本发明涉及(n-氨基正己酰氨甲环酰)-氨基正己酰-aa(式中aa为l-asp残基和l-glu残基),涉及它们的制备方法,涉及它们的抗肿瘤活性,涉及它们的抗肿瘤转移活性,以及涉及它们的抗炎活性活性,因而本发明涉及它们在制备抗肿瘤药物,抗肿瘤转移药物和抗炎药物中的应用。本发明属于生物医药领域。
背景技术
侵袭和转移过程是恶性肿瘤的基本生物学特征,是肿瘤研究工作的困境之一。癌症转移是肿瘤患者发病和死亡的主要原因,约占癌症死亡人数的90%。目前对肿瘤浸润转移的研究已从不同方面进行了深入的探讨。其中,尿激酶型纤溶酶原激活物(upa)系列在肿瘤侵袭转移中的作用成为当今研究的热点之一。尿激酶型纤溶酶原激活物(upa)系统,这是一种丝氨酸蛋白酶家族,在肿瘤的浸润转移起着至关重要的作用。该系统包括尿激酶型纤溶酶原激活物(upa)、尿激酶受体(upar)、纤溶酶原激活物抑制剂(pai),该系统涉及多种生理和病理过程,包括细胞迁移,血管生成,炎症,胚胎发育,肿瘤生长和转移。
氨基正己酸能与纤溶酶原激活物产生竞争性抑制,使纤溶酶原不能激活为纤溶酶,是临床上治疗纤溶性出血药物。氨甲环酸与纤溶酶原结合,同样能发挥抗纤溶作用。两者分别能通过阻止upa/upar相互作用和抑制纤溶酶活化来达到抑制upa系统的相关作用。氨基正己酸和氨甲环酸抑制upa系统的最低有效剂量分别为3.8mmol/kg和3.2mmol/kg。发明人认为它们的毒副作用与它们的最低有效剂量高有直接关系。发明人认识到先将两种已知的upa系统抑制剂合理组合,再连接氨基酸而构建的新型u-pa抑制剂在低剂量下就应具有抗肿瘤、抗肿瘤转移和抗炎三重作用。根据这种认识,经过3年探索,发现用酸性氨基酸(l-asp、l-glu)修饰的氨基正己酰甲环酰氨基正己酸衍生物在0.5μmol/kg剂量下不仅具有抗肿瘤转移活性,而且同时具有抗肿瘤和抗炎的活性。因为药物的毒副作用都可以随剂量降低而消失,所以有效剂量比氨基正己酸和氨甲环酸至少降低6400倍表明了这种结构修饰有突出的技术效果。根据这些发现,发明人提出了本发明。
技术实现要素:
本发明的第一个内容是提供下式的(n-氨基正己酰氨甲环酰)-氨基正己酰-aa(式中aa为l-asp残基和l-glu残基)。
本发明的第二个内容是提供(n-氨基正己酰氨甲环酰)-氨基正己酰-aa(aa为l-asp残基和l-glu残基)的合成方法,该方法包括:
(1)boc-氨甲环酸与氨基正己酸甲酯缩合得boc-氨甲环酰基氨基正己酸甲酯(1);
(2)boc-氨甲环酰基-氨基正己酸甲酯在氯化氢的乙酸乙酯溶液中脱boc得氨甲环酰基氨基正己酸甲酯(2);
(3)boc-氨基正己酸与氨甲环酰基氨基正己酸甲酯(2)缩合得(n-boc-氨基正己酰氨甲环酰)-氨基正己酸甲酯(3);
(4)化合物3皂化脱甲基得到(n-boc-氨基正己酰氨甲环酰)-氨基正己酸(4);
(5)化合物4在氯化氢的乙酸乙酯溶液中脱boc得(n-氨基正己酰氨甲环酰)-氨基正己酸(7);
(6)化合物4与aa-obzl(aa为l-asp残基和l-glu残基)缩合得(n-boc-氨基正己酰氨甲环酰)-氨基正己酰基氨基酸苄酯(5a,b)。
(7)化合物5a,b先氢解脱苄氧羰基、再在氯化氢的乙酸乙酯溶液中脱boc得到(n-氨基正己酰基氨甲环酰)-氨基正己酰-aa(6a,b)(aa为l-asp残基和l-glu残基)。
本发明的第三个内容是评价(n-氨基正己酰氨甲环酰)-氨基正己酰-aa(aa为l-asp残基和l-glu残基)抑制c57bl/6小鼠抗肺癌转移活性。
本发明的第四个内容是评价(n-氨基正己酰氨甲环酰)-氨基正己酰-aa(aa为l-asp残基和l-glu残基)对s180小鼠肿瘤生长的抑制应用。
本发明的第五个内容是评价(n-氨基正己酰氨甲环酰)-氨基正己酰-aa(aa为l-asp残基和l-glu残基)对icr小鼠炎症的抑制作用。
附图说明
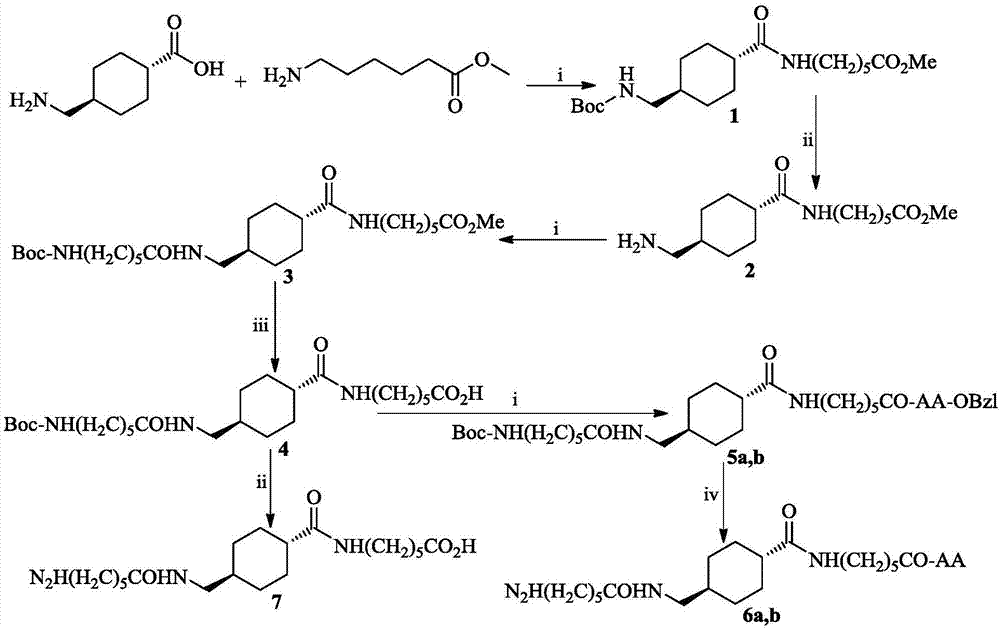
图1(n-氨基正己酰氨甲环酰)-氨基正己酰-aa的合成路线.5a和6a中aa为l-asp残基;5b和6b中aa为l-glu残基。i)二环己基碳二亚胺(dcc),1-羟基苯并三唑(hobt),n-甲基吗啉(nmm),干燥四氢呋喃(thf);ii)氯化氢的乙酸乙酯溶液(4m);iii)ch3oh,2mnaoh;iv)pd/c,h2,ch3oh;氯化氢的乙酸乙酯溶液(4m)。
具体实施方式
为了进一步阐述本发明,下面给出一系列实施例。这些实施例完全是例证性的,它们仅用来对本发明进行具体描述,不应当理解为对本发明的限制。
实施例1制备(n-boc-氨甲环酰)-氨基正己酸甲酯(1)
将0.69g(2.68mmol)boc-氨甲环酸混悬于50ml干燥四氢呋喃中,在0℃下依次加入0.66g(3.20mmol)二环己基碳二亚胺(dcc)和0.44g(3.26mmol)1-羟基苯并三唑(hobt),搅拌30min。然后加入0.49g(2.70mmol)氨基正己酸甲酯,并用n-甲基吗啉(nmm)调节溶液ph至9,室温搅拌6h,tlc(二氯甲烷/甲醇=30/1)显示反应完成。减压浓缩除去溶剂,残留物用50ml乙酸乙酯溶解,过滤。滤液先后用20ml饱和nahco3溶液洗涤3次,20ml饱和nacl溶液洗涤3次,20ml饱和khso4溶液洗涤3次,20ml饱和nacl溶液洗涤3次,20ml饱和nahco3溶液洗涤3次,20ml饱和nacl溶液洗涤3次,乙酸乙酯层用无水硫酸钠干燥12h。过滤,滤液减压浓缩至干,得到0.83g(80%)标题化合物,为无色固体。esi-ms(m/e):385[m+h]+。
实施例2制备n-氨甲环酰基-氨基正己酸甲酯·盐酸盐(2)
搅拌下将6.00g(15.62mmol)化合物1与60ml氯化氢的乙酸乙酯溶液(4m)在-10℃缓慢混合,并保持-10℃搅拌5h。tlc(二氯甲烷/甲醇=30/1)显示反应完成。减压浓缩除去溶剂,残留物用无水乙酸乙酯溶解,得到的溶液减压浓缩。该操作重复3次。固体再用无水乙醚充分悬浮,去乙醚,得到的无色固体直接用于下一步反应。esi-ms(m/e):322[m+h]+。
实施例3制备(n-boc-氨基正己酰氨甲环酰)-氨基正己酸甲酯(3)
采用实施例1的方法从3.36g(14.55mmol)boc-氨基正己酸和4.67g(14.57mmol)化合物2得到淡黄色固体。该固体用乙酸乙酯充分磨洗,得到6.20g(85%)标题化合物,为无色固体。esi-ms(m/e):498[m+h]+。
实施例4制备(n-boc-氨基正己酰氨甲环酰)-氨基正己酸(4)
将6.20g(12.47mmol)化合物3溶于20ml甲醇,0℃用naoh水溶液(2m)调节ph至12。控制0℃和ph12搅拌4h,tlc(二氯甲烷/甲醇=25/1)显示反应完成。反应混合物用饱和khso4溶液调节ph至7,减压浓缩。残留物继续用饱和khso4溶液调节ph至2,用乙酸乙酯萃取,将乙酸乙酯层合并,用饱和nacl溶液洗涤至中性。乙酸乙酯相用无水硫酸钠干燥12h。过滤,滤液减压浓缩至干,得到4.60g(76%)标题化合物,为无色固体。esi-ms(m/e):484[m+h]+。
实施例5制备(n-氨基正己酰氨甲环酰)-氨基正己酸(7)
采用实施例2的方法,从1.30g(2.69mmol)化合物4得到0.87g(84%)标题化合物,为无色固体。mp236-239℃;
实施例6制备(n-boc-氨基正己酰氨甲环酰)-氨基正己酰天冬氨酸苄酯(5a)
采用实施例1的方法,从3.00g(6.21mmol)化合物4和3.01g(6.21mmol)tos·asp(obzl)-obzl得0.90g(19%)标题化合物,为无色固体。esi-ms(m/e):779[m+h]+。
实施例7制备(n-boc-氨基正己酰氨甲环酰)-氨基正己酰谷氨酸苄酯(5b)
采用实施例1的方法,从4.00g(8.28mmol)化合物4和2.51g(6.91mmol)tos·glu(obzl)-obzl得2.33g(43%)标题化合物,为无色固体。esi-ms(m/e):793[m+h]+。
实施例8制备(n-氨基正己酰氨甲环酰)-氨基正己酰天冬氨酸(6a)
将0.81g(1.04mmol)化合物5a溶于20ml甲醇中,加入80mgpd/c,室温通10h氢气,tlc(二氯甲烷/甲醇=5/1)显示反应完成。过滤除去pd/c,滤液减压浓缩至干。残留物在-10℃与5ml氯化氢的乙酸乙酯溶液(4m)缓慢混匀,保持-10℃搅拌5h。tlc(乙酸乙酯/水/冰醋酸=3/1/1)显示反应完全。减压浓缩,残留物用无水乙酸乙酯溶解,压浓缩,残留物用无水乙酸乙酯溶解。该操作重复3次。得到的固体用无水乙醚充分悬浮,静置,去乙醚。得到的无色固体在0℃用饱和nahco3溶液调节无色固体ph至7,用c18柱层析纯化,得420mg(66%)标题化合物,为无色固体。mp263-264℃.
实施例9制备(n-氨基正己酰氨甲环酰)-氨基正己酰谷氨酸(6b)
采用实施例8的方法,从0.91g(1.15mmol)化合物5b得到285mg(0.48%)标题化合物,为无色固体。mp251-252℃.
实施例10测定化合物6a,b的抗肿瘤转移活性
本测定模型用lewis小鼠肺癌细胞(llc,购自atcc)接种,选用dmem培养基(含10%经灭活的胎牛血清,1×105u/l青霉素和100mg/l链霉素),按照贴壁细胞培养方法每两天传代一次,富集细胞。待细胞生长状态良好并处于对数生长期时消化细胞,用生理盐水调整细胞密度至1×107个/ml。胎盘蓝染色,使活细胞计数>95%。取近交系c57bl/6雄性小鼠(spf级,体重20±2g),左手固定小鼠。用75%乙醇对小鼠右前肢腋窝皮肤消毒。右手持1ml无菌注射器往小鼠腋部皮下注射llc肿瘤细胞悬液,每只小鼠注射0.2ml。小鼠接种10天后,长出直径约4-5mm的肿瘤即为瘤源。接种10天的lewis肺癌荷瘤小鼠乙醚麻醉,脱颈椎处死。用75%乙醇浸泡10min,消毒,在超净工作台上剥离瘤体。选择生长良好的肿瘤组织在无菌平皿中剪碎,置于玻璃制造的组织匀浆器内。按瘤块重比生理盐水体积为1比3(g比ml)的比例加温度为4℃的生理盐水,轻轻研磨制成细胞悬液。细胞悬液过200目细胞筛制单细胞悬液。用生理盐水调单细胞悬液的细胞密度至1.5×107个/ml。胎盘蓝染色,使活细胞计数>95%。左手固定近交系c57bl/6雄性小鼠,用75%的乙醇对小鼠右前肢腋窝皮肤消毒。右手持1ml无菌注射器于小鼠腋部皮下注射瘤细胞悬液,每只注射0.2ml。接种10天后小鼠长出直径4-5mm的肿瘤,按测得的肿瘤体积将接种小鼠随机分组。每组12只小鼠。接种肿瘤的第11天小鼠或口服公认的抗肿瘤转移肽rgds的生理盐水溶液(剂量为20μmol/kg/天)或口服化合物6a,b的生理盐水溶液(剂量为0.5μmol/kg/天)或口服化合物7的生理盐水溶液(剂量为5μmol/kg/天)或口服生理盐水(剂量为10ml/kg/天),每天给1次药,连续给药12天,每隔两天测量并记录肿瘤体积。最后一次给药的次日测量瘤体积,乙醚麻醉脱颈椎处死,取小鼠的肿瘤称重,取小鼠的肺并计算肿瘤肺部转移的瘤节数。用t检验对数据进行统计分析。结果见表1。在0.5μmol/kg剂量下化合物6a,b不仅有效地抑制肿瘤肺转移,而且活性与剂量比它们高400倍的rgds及剂量比它们高10倍的化合物7没有显著性差异。这些数据表明,本发明有显著的技术效果。
表1化合物6a-b的抗肿瘤转移活性
a)与生理盐水比p<0.01,与rgds及化合物7比p>0.05;n=12.
实施例11测定化合物6a,b的抗肿瘤生长活性
测定前将阿霉素,化合物7和化合物6a,b都用生理盐水溶解,用于s180小鼠给药。在无菌环境中取接种于雄性icr小鼠10天生长旺盛的s180腹水瘤液,用生理盐水稀释成(1:2)的液体充分混合,将肿瘤细胞悬液用新鲜配制的0.2%台盼蓝染色,混匀后按白细胞计数方法计数,染蓝色者为死细胞,不染色者为活细胞。按细胞浓度=4大方格内活细胞数/4×104×稀释倍数=细胞数/ml计算细胞密度,按细胞存活率=活细胞数/(活细胞数+死细胞数)×100%计算细胞存活率。将存活率大于90%的瘤液用匀浆法制成密度为2.0×107个/ml的细胞悬液。该细胞悬液接种于小鼠右腋皮下(0.2ml/只),制造s180荷瘤小鼠。接种24h后s180荷瘤小鼠每日腹腔注射阿霉素的生理盐水溶液(剂量为2μmol/kg/天g)或每日口服化合物7的生理盐水溶液(剂量为5μmol/kg/天)或每日口服化合物6a,b的生理盐水溶液(剂量为0.5μmol/kg/天)。每天给药一次,连续给药12天。最后一次给药的次日测量瘤体积,乙醚麻醉脱颈椎处死,然后用镊子固定小鼠右腋肿瘤生长部位,剪开皮肤钝性剥离肿瘤并称重。用瘤重(均值±sdg)表示疗效,数据用t检验和方差分析。结果见表2。在0.5μmol/kg剂量下化合物6a,b不仅有效地抑制肿瘤肺转移,而且活性与剂量比它们高10倍的化合物7没有显著性差异。这些数据表明,本发明有显著的技术效果。
表2化合物6a,b对s180小鼠肿瘤生长的影响
a)与生理盐水比p<0.01,与化合物7比p>0.05;n=12.
实施例12测定化合物6a,b的抗炎活性
因为二甲苯引起的小鼠耳肿胀被公认为急性炎症模型,所以本发明在二甲苯引起的小鼠耳肿胀模型上测定化合物6a,b的治疗作用。因为阿司匹林是治疗急性炎症的阳性药,所以本发明选择阿司匹林为阳性对照药。icr雄性小鼠(体重42±3g)在温度为22℃的环境静息2天,自由饮水和进食。之后,随机分为生理盐水组(剂量为0.2ml/只),阿司匹林组(剂量为1.11mmol/kg),化合物7组(剂量为5μmol/kg)及化合物6a,b组(剂量为0.5μmol/kg),每组12只小鼠。测定时小鼠按所在组或口服生理盐水,或口服阿司匹林,或口服化合物7,或口服化合物6a,b。给药30min后,往小鼠的左耳廓均匀涂抹30μl二甲苯,2h后小鼠接受乙醚麻醉,断颈处死,剪下左右两耳,用7mm的打孔器在两耳的相同位置取圆形耳片,称重,求出两耳肿胀差值作为肿胀度。即肿胀度=左耳圆片重量–右耳圆片重量。结果见表3。在0.5μmol/kg剂量下化合物6a,b不仅能有效地抑制二甲苯引起的小鼠耳肿胀,而且活性与剂量比它们高10倍的化合物7没有显著性差异。这些数据表明,本发明有显著的技术效果。
表3化合物6a,b对二甲苯引起的小鼠耳肿胀的影响
a)与生理盐水比p<0.01,与化合物7比p>0.05;n=12。
- 还没有人留言评论。精彩留言会获得点赞!