芳氧基取代丙‑2‑醇胺的衍生物作为β3肾上腺素能受体拮抗剂、制备方法和用途与流程
本发明属于药物合成技术领域,涉及通式i所示的含芳氧基取代丙-2-醇胺类化合物及其制备方法,体内体外的生物活性和用途,更具体的说,涉及具有以下结构的芳氧基取代丙-2-醇胺类化合物i,以及他们的制备方法,和这些化合物在治疗或预防β3-肾上腺素能受体相关联的各种疾病的应用。
背景技术:
β-肾上腺素能受体(ar)是属于g蛋白偶联受体家族中的整合膜蛋白。β1-ar和β2-ar的药理性特征已经得到了详尽的研究和描述。在组织制备中,内源性儿茶酚胺配体的大多数药理性质通过主要存在的β1-ar和β2-ar得到了合理的阐述。然而,在某些组织中,尤其是脂肪组织,β1-ar和β2-ar亚型的选择性抑制剂并没有如预测般减弱激动剂的效果,因此这些数据支持存在β3-ar这一观点。
在啮齿动物的白色脂肪组织中,β3-ar占细胞表面上β-ars的90%,这个数据是通过饱结合实验测定出来的。相比之下,通过敏感逆转录聚合酶链反应,人类β3-ar仅在人类脂肪组织、结肠和胆囊中被检测到。由于缺乏对人类可选择性的化合物,确定人体脂肪细胞内β3-ar的相对数量及其重要性就变得尤为复杂。
为了实现β3-ar在人类和灵长目动物的脂肪组织中的药理性作用和功能性表征,研发出具有高效性和选择性的抑制剂。目前典型的β3-ar抑制剂主要有两种:一类是芳氧基丙醇胺基四氢β3-ar抑制剂(sr59230a),另一类是芳氧基丙醇胺类β3-ar抑制剂(l-748,337),其结构如下所示。
这一类抑制剂中最有效的sr59230a在鼠类棕色脂肪细胞、大鼠结肠动力试验和人结肠圆形平滑肌松弛活性测定试验中都具有β3-ar的选择性。但是β3-ar拮抗剂sr59230a对人类克隆的β1-ar和β2-ar显示出更高的亲和力,因此似乎是一种有效、并非选择性的β3-ar拮抗剂。
另一类抑制剂l-748,337属于人类和恒河猴β3-ar选择性拮抗剂。l-748,337对人类β3-ar与β1-ar显示出大于90倍的选择性,对人类β2-ar分别显示出45倍的选择性。
技术实现要素:
本发明的目的是提出一类含芳氧基取代丙-2-醇胺类化合物或其可药用盐或其立体异构体或其立体异构体的可药用盐,此类化合物具有β3-肾上腺素能受体拮抗剂活性。
本发明的另一目的是提供一种含芳氧基取代丙-2-醇胺类化合物的制备方法。
本发明的再一目的在于提供一种含芳氧基取代丙-2-醇胺类化合物在治疗或预防各种与β-肾上腺素能受体相关联的疾病的应用。
本发明的目的是这样实现的:
本发明设计一种含芳氧基取代丙-2-醇胺类化合物或其可药用盐或其立体异构体或其立体异构体的可药用盐,具有化学式i所示的结构:
其中,
r1是
r2是-环己烷,-异丙基,-苯基或-ch2-苯基,且其中各自中的所述的苯基任选被一个独立地选择卤素、-och3、-nhcooch3或-nhcoc5h11的基团取代;
x是-co-,-cs-或-so2-;
y是-nh-,-ch2-或无。
所述的含芳氧基取代丙-2-醇胺类化合物包含:
或其可药用盐或其立体异构体或其立体异构体的可药用盐。
本发明提供了一种含芳氧基取代丙-2-醇胺类化合物的制备方法,该方法包括如下步骤:
其中,x、y、r1和r2的定义如前所述:
i)使化合物ii与化合物iii在还原剂条件下得到化合物iv。还原剂为硼氢化钠或氰基硼氢化钠等,溶剂为甲醇或者乙醇等,反应温度为20-80℃;
ii)化合物iv与化合物v反应,得到s型光学异构体的化合物vi。反应溶剂为2-异丙醇或乙醇等,反应温度为60-90℃;
iii)化合物vi还原生成化合物vii。还原剂为氯化亚锡、铁粉/盐酸、铁粉/氯化铵或氢氧化铁/水合肼等,反应溶剂为2-异丙醇或乙醇等,反应温度为60-90℃;
iv)在碱的条件下,化合物vii反应生成化合物viii。碱为三乙胺,吡啶,n,n-二异丙基乙胺或4-二甲氨基吡啶等,反应溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、四氢呋喃或二氯甲烷等,反应温度为0-25℃;
v)在金属催化剂的条件下,化合物viii脱苄基保护得到s型光学异构体的化学式i所示的目标化合物。金属催化剂为雷尼镍、氢氧化钯或钯碳等,反应溶剂为甲醇、n,n-二甲基甲酰胺或四氢呋喃等,反应温度为0-25℃;
r1为
1)在金属催化剂的条件下,化合物1加氢还原生产化合物2。金属催化剂为雷尼镍、氢氧化钯或钯碳等,反应溶剂为甲醇、n,n-二甲基甲酰胺、1,4-二氧六环或四氢呋喃等,反应温度为0-25℃;
2)在碱的条件下,化合物2与乙酰氯反应,得到化合物3。碱为三乙胺、吡啶、n,n-二异丙基乙胺、氢氧化钠或钠氢等,反应溶剂为二氯甲烷、n,n-二甲基甲酰胺或四氢呋喃等,反应温度为0-25℃;
3)在碱的条件下,化合物3水解得到化合物4。碱为碳酸钾、氢氧化钾或氢氧化钠等,反应溶剂为水、甲醇、四氢呋喃或1,4-二氧六环等,反应温度为0-35℃;
4)在碱的条件下,化合物4与化合物5反应,得到中间体化合物v。碱为三乙胺,碳酸钾,氢氧化钠或钠氢等,反应溶剂为水、四氢呋喃或1,4-二氧六环等,反应温度为0-25℃。
所述的化合物进行了选择性β3-肾上腺素能受体拮抗性的筛选和在3t3-l1脂细胞的体外脂解作用的筛选。
所述的化合物用于治疗通过β3-肾上腺素能受体调节的疾病或病症。
本发明提供的一种新型的含芳氧基取代丙-2-醇胺类化合物,此类化合物显示出更高的亲和力的,有效的选择性β3-ar拮抗性,并对体内和内外的脂解作用有一定抑制作用。
附图说明
图1为β3-肾上腺素能受体拮抗剂作用于肿瘤恶病质小鼠去瘤体重变化示意图;
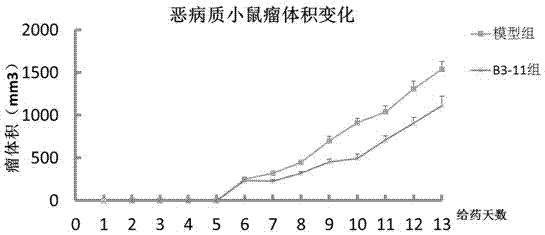
图2为β3-肾上腺素能受体拮抗剂作用于肿瘤恶病质小鼠去瘤体积变化示意图;
图3为β3-肾上腺素能受体拮抗剂作用于肿瘤恶病质小鼠后小鼠血清甘油含量示意图。
具体实施方式
本发明的含芳氧基取代丙-2-醇胺类化合物和制备方法在如下实施例中更详细的叙述,但实施例不构成对本发明的限制。
实施例1
1.1化合物2的合成
氮气氛围下,将10.0g化合物1和1g雷尼镍加入到500ml烧瓶中,加入200ml甲醇,置换氢气氛围,将体系室温反应过夜。tlc监测反应完全,反应液通过硅藻土过滤,滤液浓缩得到得化合物2为白色固体10g,产率97%。hplc分析表明纯度97%,无需进一步分离纯化直接用于下一步反应。m.p.170-172℃;esi-msm/z:124.65,[m+h]+.
实施例2
1.2化合物3的合成
将9.0g化合物3加入到250ml圆底烧瓶中,依次加入100ml二氯甲烷、11.8ml吡啶和11.9ml乙酰氯,将体系室温反应2小时。tlc监测反应完全后,反应体系用饱和氯化铵溶液和饱和食盐水洗涤,无水硫酸钠干燥。旋干溶剂后得化合物3为黄色油状物13.4g,收率85.0%。hplc分析表明纯度97%,无需进一步分离纯化直接用于下一步反应。esi-msm/z:208.26,[m+h]+.
实施例3
1.3化合物4的制备
将13.4g化合物3加入到250ml烧瓶中,依次加入75ml质量分数为15%的氢氧化钠溶液和75ml甲醇,将体系室温反应16小时。tlc监测反应完全,减压除去有机溶剂,加入75ml2m盐酸调ph=4。水相用乙酸乙酯萃取(100ml×3),合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥。旋干溶剂后的粗产品,经柱层析(二氯甲烷:甲醇=20:1)分离纯化得化合物4为亮黄色固体9.8g,收率92%。m.p.95-97℃;esi-msm/z:165.90,[m+h]+.
1hnmr(400mhz,dmso)δ9.31(s,1h),8.28(s,1h),7.09(t,j=7.9hz,1h),6.65(d,j=4.5hz,2h),6.62(d,j=8.4hz,1h),4.16(d,j=5.9hz,2h),1.86(s,3h).
13cnmr(101mhz,dmso)δ169.03,157.32,140.95,129.16,117.79,114.04,113.63,41.99,22.51.
实施例4
1.4化合物v的制备
将5.0g化合物4和1.5g氢氧化钠加入到500ml烧杯中,加入5ml水和20ml1,4-二氧六环,将体系室温反应5小时,在室温条件下加入3.6ml化合物5,将体系室温反应24小时。向反应体系中加入30ml水,用乙酸乙酯萃取(50ml×3),合并有机相,用饱和碳酸氢钠溶液、和饱和食盐水洗涤,无水硫酸钠干燥。旋干溶剂后的粗产品,经柱层析(二氯甲烷:甲醇=40:1)分离纯化得化合物viii为淡黄色固体3.3g,收率49.6%。m.p.73-75℃;esi-msm/z:222.56,[m+h]+.
1hnmr(400mhz,cdcl3)δ7.24(t,j=7.8hz,1h),6.87(d,j=7.7hz,1h),6.83(d,j=11.2hz,2h),5.95(s,1h),4.38(d,j=5.8hz,2h),4.23(dd,j=11.1,2.9hz,1h),3.92(dd,j=11.0,5.8hz,1h),3.34(td,j=6.2,3.1hz,1h),2.90(t,j=4.5hz,1h),2.75(dd,j=4.9,2.6hz,1h),2.01(s,3h).
实施例5
1.5化合物iv的制备
将10.0g化合物ii和2.1g氢氧化钠加入到250ml烧瓶中,加入30ml水和50ml乙酸乙酯,将体系室温反应1小时,用乙酸乙酯萃取(30ml×3),合并有机相,用饱和食盐水洗涤,浓缩得到粗品8.2g。
氮气氛围下,将8.2g粗品和5.7ml化合物iii加入到250ml烧瓶中,加入70ml乙醇,将体系升温回流反应16小时。反应液冷却至0℃,缓慢加入2.05g硼氢化钠,将体系室温反应2小时。tlc监测反应完全,加入50ml质量分数为20%盐酸溶液保持体系ph为4,析出白色固体。过滤得到化合物iv为白色固体14.0g,收率97%。m.p.255-257℃;esi-msm/z:293.76,[m+h]+.
1hnmr(400mhz,dmso)δ9.74(s,2h),8.20(d,j=8.7hz,2h),7.60(dd,j=7.2,1.7hz,2h),7.56(d,j=8.7hz,2h),7.42(d,j=6.0hz,3h),4.17(s,2h),3.20(s,4h).
13cnmr(101mhz,dmso)δ146.38,145.66,132.04,130.13,130.01,128.74,128.51,123.66,49.77,46.65,31.01.
实施例6
1.6化合物vi的制备
将6.0g化合物iv和940mg氢氧化钠加入到250ml烧瓶中,加入30ml水和50ml乙酸乙酯,将体系室温反应1小时,用乙酸乙酯萃取(25ml×3),合并有机相,用饱和食盐水洗涤,浓缩得到粗品5.2g。
将5.2g粗品和4.0g化合物v加入到100ml烧瓶中,加入25ml2-异丙醇,将体系升温回流反应16小时。tlc监测反应完全,反应液浓缩得到粗品。粗产品经柱层析(二氯甲烷:甲醇=40:1)分离纯化得化合物vii为黄色油状物8.5g,收率98.0%。esi-msm/z:478.56,[m+h]+.
1hnmr(400mhz,dmso)δ8.30(t,j=5.7hz,1h),8.06(d,j=8.5hz,2h),7.41(d,j=8.7hz,2h),7.30–7.20(m,5h),7.18(t,j=7.8hz,1h),6.80(d,j=7.5hz,1h),6.71(s,1h),6.65(dd,j=8.2,2.0hz,1h),4.83(d,j=4.9hz,1h),4.21(d,j=5.9hz,2h),3.87(dd,j=9.3,5.0hz,1h),3.82–3.73(m,2h),3.71–3.55(m,2h),2.89(t,j=7.0hz,2h),2.83–2.69(m,2h),2.65(dd,j=13.2,6.7hz,1h),2.54(d,j=5.8hz,1h),1.87(s,3h).
13cnmr(101mhz,dmso)δ169.06,158.66,149.30,145.69,141.12,139.19,129.91,129.16,128.71,128.02,126.75,123.12,119.28,113.46,112.24,70.49,67.22,58.41,56.28,55.10,41.99,32.38,22.53.
实施例7
1.7化合物vii的制备
将6.0g化合物vi和8.5g氯化亚锡加入到500ml烧瓶中,加入200ml乙醇,将体系升温回流反应2小时。tlc监测反应完全,加入3m的氢氧化钠溶液保持体系ph值为9,反应液通过硅藻土过滤,减压除去溶剂乙醇,向反应体系中加入50ml水,用乙酸乙酯萃取(50ml×3),合并有机相,用饱和食盐水洗涤,无水硫酸钠干燥。旋干溶剂后的粗产品,经碱性氧化铝柱层析(二氯甲烷:甲醇=10:1)分离纯化得化合物vii为黄色油状物5.4g,收率96%。esi-msm/z:447.60,[m+h]+.
1hnmr(400mhz,dmso)δ8.31(t,j=5.7hz,1h),7.28(d,j=4.5hz,4h),7.21(dd,j=10.4,5.3hz,2h),6.86–6.68(m,5h),6.44(d,j=8.2hz,2h),4.85–4.69(m,3h),4.21(d,j=6.0hz,2h),3.95–3.84(m,2h),3.73(dd,j=11.3,6.7hz,2h),3.61(d,j=13.8hz,1h),2.69–2.51(m,6h),1.87(s,3h).
13cnmr(101mhz,dmso)δ169.06,158.77,146.41,141.10,139.53,129.24,128.89,128.62,128.00,127.17,126.66,119.25,113.89,113.49,112.46,70.61,67.26,58.58,56.49,42.01,31.84,22.53.
实施例8
1.8化合物viii的制备
氮气氛围下,将2.0g化合物vii、800mg化合物6和1.1ml吡啶加入到100ml烧瓶中,加入30ml二氯甲烷,将体系室温反应过夜。tlc监测反应完全,反应液通过硅藻土过滤,用饱和氯化铵溶液和饱和食盐水洗涤,无水硫酸钠干燥。旋干溶剂后的粗产品,经柱层析(二氯甲烷:甲醇=20:1)分离纯化得化合物viii为白色固体1.9g,收率75.0%。
m.p.70-72℃;esi-msm/z:567.72,[m+h]+.
实施例9
1.9化合物i的制备
氮气氛围下,将1.8g化合物vii和100mg雷尼镍加入到500ml烧瓶中,加入200ml甲醇,置换氢气氛围,将体系室温反应过夜。tlc监测反应完全,反应液通过硅藻土过滤,滤液浓缩得到粗品。粗产品经柱层析(二氯甲烷:甲醇=20:1)分离纯化得化合物i为白色固体1.0g,收率66.1%。m.p.203-205℃;esi-msm/z:477.5900,[m+h]+.
1hnmr(400mhz,dmso)δ9.45(d,j=3.6hz,2h),9.18(s,1h),8.94(s,1h),8.43(s,1h),7.45(dd,j=11.4,8.4hz,4h),7.25(dd,j=16.9,8.2hz,4h),7.16(d,j=8.0hz,2h),6.94(t,j=7.2hz,1h),6.89–6.78(m,3h),4.22(d,j=5.4hz,3h),3.97(s,2h),3.28–3.11(m,3h),3.09–2.87(m,3h),1.88(s,3h).
13cnmr(101mhz,dmso)δ169.16,158.27,152.75,141.28,139.94,138.62,130.13,129.32,128.89,128.69,121.49,119.73,118.07,117.80,113.57,112.59,69.64,64.86,49.55,48.33,41.96,30.72,22.53.
实施例10
选择性β3-肾上腺素能受体拮抗性化合物的筛选
构建稳定表达β1ar受体和gα16基因的hek293细胞株(hek293/beta1ar/gα16)。简单步骤如下:将β1ar表达载体和gα16基因表达载体通过电转方式导入到hek93细胞中。利用抗生素杀死没有转染上述表达载体的细胞,具有抗性的细胞则会以克隆形式生长。挑选这些具有抗性的克隆独立培养,最后获得稳定表达β1ar和gα16基因的hek93细胞株。
构建稳定表达β2ar受体和gα16基因的hek293细胞株(hek293/beta2ar/gα16)。简单步骤如下:将β2ar表达载体和gα16基因表达载体通过电转方式导入到hek93细胞中。利用抗生素杀死没有转染上述表达载体的细胞,具有抗性的细胞则会以克隆形式生长。挑选这些具有抗性的克隆独立培养,最后获得稳定表达β2ar和gα16基因的hek93细胞株。
构建稳定表达β3ar受体和gα16基因的hek293细胞株(hek293/beta3ar/gα16)。简单步骤如下:将β3ar表达载体和gα16基因表达载体通过电转方式导入到hek93细胞中。利用抗生素杀死没有转染上述表达载体的细胞,具有抗性的细胞则会以克隆形式生长。挑选这些具有抗性的克隆独立培养,最后获得稳定表达β3ar和gα16基因的hek93细胞株。
利用β1ar/gα16/hek293稳转细胞株进行化合物的钙流筛选实验。将稳定表达beta1ar/gα16/hek293细胞株接种在96孔板里,培养24小时后,将细胞与2μmfluo4-am(购自invitrogen)染料在37℃孵育45分钟,移去染料,加入50μl含有待测化合物的hbss(5.4mmkcl,0.3mmna2hpo4,0.4mmkh2po4,4.2mmnahco3,1.3mmcacl2,0.5mmmgcl2,0.6mmmgso4,137mmnacl,5.6mmd-glucose和250μmsulfinpyrazone),室温孵育10分钟,然后用flexstation3微孔板检测仪(moleculardevices)加入25μl含有isopreterenol的hbss进行刺激,同时用485nm的光激发并于525nm波段检测细胞
利用β2ar/gα16/hek293稳转细胞株进行化合物的钙流筛选实验。将稳定表达β2ar/gα16/hek293细胞株接种在96孔板里,培养24小时后,将细胞与2μmfluo4-am(购自invitrogen)染料在37℃孵育45分钟,移去染料,加入50μl含有待测化合物的hbss(5.4mmkcl,0.3mmna2hpo4,0.4mmkh2po4,4.2mmnahco3,1.3mmcacl2,0.5mmmgcl2,0.6mmmgso4,137mmnacl,5.6mmd-glucose和250μmsulfinpyrazone),室温孵育10分钟,然后用flexstation3微孔板检测仪(moleculardevices)加入25μl含有isopreterenol的hbss进行刺激,同时用485nm的光激发并于525nm波段检测细胞
利用β3ar/gα16/hek293稳转细胞株进行化合物的钙流筛选实验。将稳定表达β3ar/gα16/hek293细胞株接种在96孔板里,培养24小时后,将细胞与2μmfluo4-am(购自invitrogen)染料在37℃孵育45分钟,移去染料,加入50μl含有待测化合物的hbss(5.4mmkcl,0.3mmna2hpo4,0.4mmkh2po4,4.2mmnahco3,1.3mmcacl2,0.5mmmgcl2,0.6mmmgso4,137mmnacl,5.6mmd-glucose和250μmsulfinpyrazone),室温孵育10分钟,然后用flexstation3微孔板检测仪(moleculardevices)加入25μl含有isopreterenol的hbss进行刺激,同时用485nm的光激发并于525nm波段检测细胞
使用上述生物鉴定法,表1显示了选定的本发明化合物在人类β1、β2、β3-肾上腺素能受体的拮抗性。化合物编号对应于具体形式中的化合物编号。活性指定为“a”的化合物提供的ec50≤10nm;活性指定为“b”的化合物提供的ec50为10-100nm;活性指定为“c”的化合物提供的ec50为100-1000nm;活性指定为“d”的化合物提供的ec50为1000-10000nm。效价(nm);效能(ec50)
表1
实施例11
β3-肾上腺素能受体拮抗剂在3t3-l1脂细胞的体外脂解抑制作用
诱导分化3t3-l1成熟脂肪细胞。利用3t3-l1脂肪细胞建立iso刺激脂解模型:3t3-l1成熟脂肪细胞孵育含有不同浓度iso的无酚红无血清dmem培养液(含2%fattyacid-freebsa),作用2hr;培养液12000g离心5min,取上清,利用甘油检测试剂盒检测甘油含量,结果证明1μm的iso具有明显的刺激脂解作用。在iso(1μm)脂解模型上,筛选β3-肾上腺素能受体拮抗剂对脂解的抑制作用:10μmb3aa系列化合物与iso共孵育2hr(无酚红无血清dmem培养液,含2%fattyacid-freebsa),培养液12000g离心5min,取上清,利用甘油检测试剂盒检测甘油含量(μmol/l),结果指定为“+”的甘油释放量为≤10μmol/l;活性指定为“++”的甘油释放量为10-100μmol/l,结果如表2所示。
表2
实施例12
β3-肾上腺素能受体拮抗剂治疗肿瘤恶病质小鼠
动物实验方案如表3:
表3
注:健康对照组(未接种肿瘤),b3-11组(蓖麻油/dmso1:1)
取新鲜小鼠c26实体肿瘤组织,每1g肿瘤组织中加入适量生理盐水研磨后稀释成浓度约1×107/ml的单细胞悬液,每只babl/c(雄性,22g左右)小鼠腋下注射c26结肠癌细胞悬液0.1ml,对照组每只小鼠腋下注射0.1ml理盐水;接种c26肿瘤后,b3-11组每天经尾静脉给药,模型组每天腹腔注射等体积生理盐水;实验历时13天,每天记录小鼠的体重(去瘤体重如图1)、肿瘤体积(如图2)。
实验结束,收取小鼠血清,进行生化分析检测甘油含量(见图3)。
实验结果发现10天后β3-肾上腺素能受体拮抗剂在肿瘤恶病质的动物模型中可以减少恶病质的体重降低,并且还减少肿瘤的肿瘤。对照组b3-11组小鼠血清中甘油含量明显低于模型组小鼠,实验结果说明β3-肾上腺素能受体拮抗剂对于肿瘤恶病质小鼠体内的脂解有一定的抑制作用。
- 还没有人留言评论。精彩留言会获得点赞!