包含内部标记引物的核酸的测序、扩增和检测方法与流程
本申请为国际申请日2012年5月4日、国际申请号pct/ep2012/058259于2013年11月6日进入中国国家阶段、申请号201280022096.0、发明名称“包含内部标记引物的核酸的测序、扩增和检测方法”的分案申请。
本发明属于生物学和化学领域,特别是分子生物学领域。更具体来说,本发明涉及用于核酸测序或扩增和检测的方法和试剂盒。
背景技术:
如今,分子方法在诊断学和鉴定个体,例如在法医学领域或亲子鉴定中发挥重要作用。靶区域的扩增是这些方法中的中心程序。此外,方法包括在大多数情况下通过凝胶电泳如毛细管电泳来分析被扩增靶区域的长度。被扩增材料的分析通过荧光标记物的检测来进行。将标记物附连到用于扩增的位点特异性引物的5’-末端。然而,当分析扩增产物时,小的干扰性人为峰、即所谓的“染料污迹(dyeblobs)”的出现,导致基线具有高的“背景噪音”,这干扰了方法的灵敏度。“染料污迹”是电泳图中不代表特异性扩增产物,而是代表包含标记物的降解产物的峰。造成它们的分子是游离的荧光染料标记物以及与标记物相连的1-8bp长的寡核苷酸。后者可能代表降解的引物或降解的pcr产物。
现在,本发明人出人意料地发现,内部标记的寡核苷酸引物的使用减少了染料污迹的出现。因此,本申请提供了用于测序或扩增和检测核酸的方法,所述方法包括使用内部标记的核酸引物。使用内部标记的引物的技术效果是提供了具有更高灵敏度的方法。
技术实现要素:
因此,本发明解决了上面提到的问题,并提供了用于测序或扩增和检测核酸的方法,所述方法包括下列步骤:
i)提供至少一种核酸引物,其包含至少一个标记的核苷酸,
ii)提供酶和试剂,其使用所述至少一种标记的核酸引物来扩增靶核酸,
iii)将反应组分在适合于靶核酸引物的扩增的条件下温育,
iv)通过标记的核苷酸来检测扩增的核酸,
其中所述核酸引物包含下列序列:
5’-naxnb-3’,
其中n可以是任意核苷酸或修饰的核苷酸,
其中a和b表示核苷酸数量,并可以在1至150的范围内;
并且其中x是所述至少一个标记的核苷酸。
在本文中,“核酸引物”是指适用于扩增的寡核苷酸。本领域技术人员应该理解,为了适用于扩增,3’-末端应该可以通过聚合酶进行延伸。核酸引物可以使用任何适合的方法来制备,例如磷酸三酯和磷酸二酯方法或其自动化实施方式。在一种这样的自动化实施方式中,使用二乙基-亚磷酰胺作为起始原料,其可以如beaucage等,tetrahedronletters,22:1859-1862(1981)所述进行合成,该文献通过引用并入本文。一种在修饰的固体支持物上合成寡核苷酸的方法描述在美国专利号4,458,006中,所述专利通过引用并入本文。还可以使用从生物样品(例如限制性内切酶消化物)分离的引物。核酸引物的长度可以各不相同。长度可以适应于需要。
在本发明的方法的情形中,“核酸”是指样品中特定类型的核酸,尤其是rna、dna、cdna(互补dna)、lna(锁核酸)、mrna(信使rna)、mtrna(线粒体的)、rrna(核糖体rna)、trna(转运rna)、nrna(核rna)、sirna(短干扰rna)、snrna(小核rna)、snorna(小核仁rna)、scarna(小卡哈尔体特异性rna)、microrna、dsrna(双链rna)、核酶、核糖开关、病毒rna、dsdna(双链dna)、ssdna(单链dna)、质粒dna、粘粒dna、染色体dna、病毒dna、mtdna(线粒体dna)、ndna(核dna)、或sndna(小核dna)等,或可以与样品中主体核酸区分开的任何其他类型或亚类的核酸。
本发明人发现,使用内部标记的核酸引物导致引起染料污迹的片段减少。如果扩增产物在检测之前必须按照其长度进行分离,则染料污迹是特别不想要的。因此,在本发明方法的一种实施方式中,步骤iv)优选地在通过标记的核苷酸检测扩增的核酸之前,包括按照长度分离扩增的核酸的步骤。本领域技术人员了解按照核酸长度分离核酸的大量方法。一种常用方法是凝胶电泳。因此,在本发明的一种实施方式中,使用凝胶电泳对扩增的核酸按照其大小进行分离。凝胶电泳是使用施加于凝胶基质的电场来分离核酸分子的技术。术语“凝胶”是指用于包含、然后分离靶分子的基质。在大多数情况下,凝胶是交联聚合物,其组成和孔隙度根据待分析的靶的具体重量/长度和组成来选择。当分离小核酸(dna、rna或寡核苷酸)时,凝胶通常由不同浓度的丙烯酰胺和交联剂构成,产生不同网孔尺寸的聚丙烯酰胺网状物。丙烯酰胺可以是例如丙烯酰胺、直链聚丙烯酰胺、聚二甲基丙烯酰胺或聚异丙基丙烯酰胺。当分离较大核酸(大于几百碱基)时,可以选择纯化的琼脂糖作为基质。在两种情形中,凝胶形成固体但有孔的基质。琼脂糖由没有交联的不带电荷的糖类的无分支长链构成,产生具有大孔的凝胶,允许分离大分子和大分子复合物。因此,在本发明的一种实施方式中,用于分离核酸的基质是聚丙烯酰胺或琼脂糖。
凝胶电泳使用在法医学、分子生物学、遗传学、微生物学和生物化学中。通过用成像装置将凝胶中标记的核酸可视化,可以对结果进行定量分析,所述成像装置通常包含激发标记物荧光的光源,例如激光。用检测装置记录来自于标记物的信号,并测量条带或斑点的强度。测量和分析大多使用专门的软件来进行,所述软件将检测到的信号按照核酸的长度和被检测核酸的量转变成电泳图。在本发明的优选实施方式中,扩增的核酸的分离通过毛细管凝胶电泳来进行。
此外,本领域技术人员了解在内部位置标记寡核苷酸的方法。一种可能方法是将标记物附连于核碱基,例如核碱基的环外氨基(在本文中也称为胺基),另一种可能方法是将标记物附连于糖的2’-碳。
“环外”是指位于环状化学结构的环外部的基团,例如环的一部分取代基对于环来说是环外的。当在本文中使用时,环外胺基是指作为杂环碱基的环的取代基的胺基,并包括胺基的氮直接附连于环结构的成员的实施方式,还包括胺基的氮可以通过居间基团连接到杂环碱基的环结构的实施方式。
此外,可以将标记物连接到2’-修饰的核苷酸,例如,将胺基直接附连于糖的2’-碳,或者胺基的氮可以通过居间基团连接到糖的2’-碳。
在本发明的一种实施方式中,将标记物通过环外胺基连接到所述被标记核苷酸(x)或连接到所述被标记核苷酸(x)的修饰的2’-位置。在优选实施方式中,将标记物连接到被标记核苷酸(x)的环外氨基。在优选实施方式中,将标记物连接到胸腺嘧啶的环外胺基。
标记寡核苷酸的可能方法对于专业技术人员来说是已知的,包括但不限于氨基-修饰物-c6’-dtcep(例如5-[n-(三氟乙酰基氨基己基)-3-丙烯酰亚胺基],也称为5-[e-2-[n-[6-(三氟乙酰胺基)己基]甲酰胺基]乙烯基])、氨基-修饰物-c2-dtcep、5-氨基烯丙基-ducep、氨基-修饰物-c6-dacep、氨基-修饰物-c6-dgcep(例如3'-o-[(二异丙基氨基)(2-氰基乙氧基)膦基]-5'-o-(4,4'-二甲氧基三苯甲基)-n2-[(二甲基氨基)亚甲基]-n8-[6-(三氟乙酰基氨基)己基]-2'-脱氧鸟苷)、氨基-修饰物-15-dtcep(例如3’-o-[(二异丙基氨基)(2-氰基乙氧基)膦基]-5’-o-(4,4’-二甲氧基三苯甲基)-5-[e-2’-[n-[15-(三氟乙酰胺基)-7-氮杂-10,13-二氧杂-8-氧基十五烷基]甲酰胺基]乙烯基]-2’-脱氧尿苷、2’-o-氨基连接物-ucep(例如3'-o-[(二异丙基氨基)(2-氰基乙氧基)膦基]-5'-o-(4,4'-二甲氧基三苯甲基)-2'-o-2-[2-(三氟乙酰胺基)乙氧基]乙基尿苷)和2'-氨基-d-尿苷。
标记的核苷酸(x)在核酸引物中的位置可以变化。然而,由于本发明人发现内部标记的核酸引物减少电泳图中染料污迹的出现,因此本发明的核酸引物被内部标记,即标记的核苷酸不是在核酸引物的5’-或3’-位置处的核苷酸。因此,在本发明的核酸引物中,“a”和“b”至少为1。“a”和“b”的值取决于标记的核苷酸的内部位置和核酸引物的总长度。在优选实施方式中,“a”和/或“b”在1至150之间,优选地在2至50之间,甚至更优选地在3至25之间,更优选地在4至17之间。在其他实施方式中,“a”在1至150之间,优选地在2至50之间,甚至更优选地在3至25之间,更优选地在4至17之间。在其他实施方式中,“b”在1至150之间,优选地在2至50之间,甚至更优选地在3至25之间,更优选地在4至17之间。本发明人出人意料地发现,标记的核苷酸越处于引物的中央位置中,减少染料污迹的效果越强。因此,在一种实施方式中,“a”和“b”相差15或更小,优选地相差10或更小,更优选地相差5或更小。在其他优选实施方式中,“a”和“b”具有相同的值。本发明人发现,当“a”和“b”为10时,减少染料污迹的效果达到最高。因此,在本发明的一种实施方式中,“a”和“b”为10。然而,专业技术人员可以根据引物的长度和序列改变标记物的确切位置。本领域普通技术人员将会理解,在整个分子中,核苷酸或修饰的核苷酸(n)不必是相同的,例如,如果“a”和“b”为5,这不意味着在得到的序列nnnnnxnnnnn中所有的“n”都是相同核苷酸。此外,专业技术人员将根据待扩增的靶核酸来选择核酸引物的核苷酸或修饰的核苷酸,即他将选择与靶核酸的一部分互补的序列。
可以根据需要并根据所需的特异性来改变核酸引物的长度,例如,较长引物具有较高特异性程度。在本发明的优选实施方式中,核酸引物具有约3至300nt之间,更优选地约5至100nt之间,甚至更优选地约6至50nt之间的总长度。在其他优选实施方式中,核酸引物具有15至34nt之间的总长度。
通过本发明的方法实现的技术效果,可以在大量不同标记物上观察到。因此,它原则上可以使用适合于标记核酸的任何标记物来进行。然而,在本发明的一种实施方式中,标记物选自由荧光团、生色团、放射性同位素、化学发光物和酶构成的标记物组。在优选实施方式中,标记物是荧光团,其优选地选自包含下列荧光团的荧光团组:5-或6-羧基荧光素(famtm),victm,nedtm,荧光素,荧光素异硫氰酸酯(fitc),ird-700/800,花青染料例如cy3tm,cy5tm,cy3.5tm,cy5.5tm,cy7tm,氧杂蒽,6-羧基-2’,4’,7’,4,7-六氯荧光素(hex),6-羧基-1,4-二氯-2’,7’-二氯-荧光素
在一种实施方式中,本发明的核酸引物包含修饰的核苷酸。修饰的核苷酸涵盖:作为标记的核苷酸的核苷酸,其选自包含上文中列出的标记物的核苷酸;修饰的核苷酸;脱碱基位点;和淬灭剂。专业技术人员能够根据需要从大量不同核苷酸类型中选出适合的核苷酸。在优选实施方式中,每个n独立地选自胸苷、腺苷、鸟苷、胞苷、尿苷和次黄苷。还涵盖核苷酸碱基,其中所述核苷酸碱基是例如次黄嘌呤、黄嘌呤、7-甲基鸟嘌呤、黄嘌呤核苷(xanthinosine)、7-甲基鸟苷、5,6-二氢尿嘧啶、5-甲基胞嘧啶、假尿苷、二氢尿苷、5-甲基胞苷。因此,在一种实施方式中,一个或多个“n”是修饰的核苷酸。
在本文中,“修饰的核苷酸”是指非天然核苷酸。“修饰的核苷酸”为本领域技术人员所公知。然而,在本发明的一种实施方式中,修饰的核苷酸选自锁核酸(lna)或肽核酸(pna)、如上文中所列出的标记的核苷酸、连接到淬灭剂的核苷酸、和脱碱基位点。
“淬灭”是指降低给定物质的荧光强度的任何过程。在本发明的情形中,“淬灭剂”是指适合于淬灭例如标记物的基本荧光而不激活的残基。各种不同过程可以导致淬灭,例如激发态反应、能量转移、复合物形成和碰撞引起的淬灭。因此,淬灭通常严重依赖于压力和温度。分子氧和碘离子是常见的化学淬灭剂。淬灭为非即时光谱测定法例如激光诱导的荧光提出了问题。淬灭是荧光共振能量转移(fret)测定法的基础。与特异性分子生物学靶相互作用后的淬灭和解淬灭,是用于分子成像的可激活光学造影剂的基础。存在几种不同的能量转移机制:两种染料即供体和受体之间的非辐射性机制(没有光子吸收或发射),
在本发明的情形中,“脱碱基位点”(也称为ap位点(脱嘌呤/脱嘧啶位点))是核酸/寡核苷酸中既没有嘌呤也没有嘧啶碱基的位置。脱碱基位点已在“用于提高杂交和引发特异性的组合物和方法”(compositionsandmethodsforenhancinghybridizationandprimingspecificity)(us-b16,361,940)和“用于杂交dna靶的无标记物检测的方法”(methodforlabel-freedetectionofhybridizeddnatarget)(us-b16,579,680)以及“使用含有脱碱基位点的dna链在水中进行核碱基识别”(useofabasicsite-containingdnastrandsfornucleobaserecognitioninwater,yoshimoto等,jacscommunications,网络版,07/04/2003)中提到。
此外,专业技术人员将会认识到,本发明的核酸引物可以由不同类型的核酸或修饰的核酸构成。因此,在优选实施方式中,核酸引物由dna、rna、pna或lna构成。
本发明人出人意料地发现,如果通过连接物将标记物附连于核酸引物内的核苷酸,可以进一步增强本发明的方法的技术效果。因此,在一种实施方式中,通过连接物将标记物附连于核苷酸。适合的连接物可以包括从现有技术已知的间隔物。例如,根据本发明,可以在标记物与核苷酸之间使用包含2至18个碳原子的连接物例如c5-连接物或c6-连接物。优选地,间隔物是支链或直链氨基连接物。在其他优选实施方式中,间隔物选自c2-至c18–连接物或氨基连接物,优选地,间隔物选自c3-至c12-连接物或氨基连接物。连接物或氨基连接物优选地是直链烷基。在非常特殊的实施方式中,间隔物是c5-间隔物或c6-间隔物。可以使用氨基连接物将伯氨基并到寡核苷酸的5’-末端、3’-末端、环外氨基或修饰的2’-位置上。可以使用活性氨基,用标记试剂来标记寡核苷酸,例如采取n-羟基琥珀酰亚胺酯的形式。
本发明的方法包括核酸的扩增。扩增可以通过各种已知扩增方法来进行。因此,在本发明的一种实施方式中,扩增通过选自下列的方法来进行:聚合酶链反应(pcr),等温扩增(例如在walker等,“链置换扩增—一种等温体外dna扩增技术”(stranddisplacementamplification--anisothermal,invitrodnaamplificationtechnique),nucleicacidsres.20(7):1691-6(1992)中),连接酶链反应(lcr;例如在landegren等,“连接酶介导的基因检测技术”(aligase-mediatedgenedetectiontechnique),science241:1077-1080,1988或wiedmann等,“连接酶链反应(lcr)—概述和应用”(ligasechainreaction(lcr)--overviewandapplications),《pcr方法和应用》(pcrmethodsandapplications)(coldspringharborlaboratorypress,coldspringharborlaboratory,ny,1994)pp.s51-s64.)中),基于转录的扩增系统(tas),基于核酸序列的扩增(nasba;kievits等,(1991)jvirolmethods35:273-286),滚环扩增(rca;例如在liu等,“滚环dna合成:小的环状寡核苷酸作为dna聚合酶的有效模板”(rollingcirclednasynthesis:smallcircularoligonucleotidesasefficienttemplatesfordnapolymerases),j.am.chem.soc.118:1587-1594(1996)中),转录介导的扩增(tma;vuorinen等,(1995)jclinmicrobiol33:1856-1859),自持序列复制(3sr),qβ扩增,链置换扩增(sda)(walker等,(1992)nucleicacidsres20(7):1691-6),多重置换扩增(mda)(dean等,(2002)procnatlacadsciusa99(8):5261-5266),限制性酶辅助的rca(wang等,(2004)genomeres14:2357–2366),单引物等温扩增(spia;dafforn等,(2004)biotechniques37(5):854-7),环介导的等温扩增(lamp;notomi等,(2000)nucleicacidsres28(12):e63),转录介导的扩增(tma),解旋酶依赖性扩增(hda),热稳定hda(thda)(an等,(2005)jbiolchem280(32):28952-28958),smart扩增方法(smap;mitani等,(2007)natmethods4(3):257-62)),定量实时pcr(qpcr),反转录酶pcr(rt-pcr),sanger测序。
本发明还涉及用于测序或扩增和检测核酸的试剂盒,所述试剂盒包含:
-包含至少一个标记的核苷酸的核酸引物,
其中所述核酸引物包含下列序列:
5’-naxnb-3’,
其中n可以是任意核苷酸或修饰的核苷酸,
其中a和b表示核苷酸数量并且可以在1至150的范围内,并且其中x是所述至少一个标记的核苷酸。
如上所概述的用于本发明方法的核酸引物的实施方式,也适用于本发明试剂盒的核酸引物。
如上所概述的,扩增方法可以选自不同方法。取决于模板核酸和扩增方法,可以将不同酶或试剂合并到本发明的试剂盒中。在其他实施方式中,除了核酸引物之外,试剂盒还包含缓冲剂、dntp和/或ntp和/或酶。
在本发明的一种实施方式中,试剂盒还包含聚合酶。聚合酶可以作为独立的反应试剂存在于试剂盒中,或者可以以合并的测定混合物例如主混合物的形式与其他试剂盒组分预先混合在一起。dna聚合酶优选地是“热稳定的”,意味着它在用于变性dna链的高温下一般是稳定的。更具体来说,热稳定dna聚合酶在pcr中使用的高温下不显著失活。这样的温度随着大量反应条件,包括ph、盐浓度和本领域已知的其他条件而变。在本发明的情形中,优选的实施方式包含可能具有5’-3’外切酶活性的热稳定dna聚合酶或具有降低的或不具有5’-3’外切酶活性的热稳定dna聚合酶。在另一种实施方式中,热稳定dna聚合酶可能另外具有3’-5’外切酶(校正读码)活性。在本领域中已报道了大量热稳定dna聚合酶。特别有用的聚合酶是从各种栖热菌属(thermus)细菌物种例如水生栖热菌(thermusaquaticus)、thermusfiliformis、黄栖热菌(thermusflavus)或嗜热栖热菌(thermusthermophilus)获得的聚合酶。其他有用的热稳定聚合酶从各种其他微生物来源包括thermococcusliteralis、强烈炽热球菌(pyrococcusfuriosus)、热袍菌属菌种(thermotogasp.)以及在wo-a-89/06691(1989年7月27日出版)中描述的微生物获得。这样的聚合酶包括但不限于嗜热栖热菌(tth)dna聚合酶、水生栖热菌(taq)dna聚合酶、thermotoganeopalitana(tne)dna聚合酶、海栖热袍菌(thermotogamaritima)(tma)dna聚合酶、thermococcuslitoralis(tli或vent.(tm).)dna聚合酶、thermuseggertssonii(teg)dna聚合酶、强烈炽热球菌(pfu)dna聚合酶、deepvent.dna聚合酶、pyrococcuswoosii(pwo)dna聚合酶、pyrococcusspkdd2(kod)dna聚合酶、嗜热脂肪芽孢杆菌(bacillussterothermophilus)(bst)dna聚合酶、bacilluscaldophilus(bea)dna聚合酶、嗜热硫化叶菌(sulfolobusacidocaldarius)(sac)dna聚合酶、嗜酸热原体(thermoplasmaacidophilum)(tac)dna聚合酶、thermusflavus(tfl/tub)dna聚合酶、红色栖热菌(thermusruber)(tru)dna聚合酶、thermusbrockianus(dynazyme)dna聚合酶、methanobacteriumthermoautotrophicum(mth)dna聚合酶、分枝杆菌dna聚合酶(mtb,mlep),及其突变体、变体和衍生物,包括在室温下无活性并且通过在高温下温育可以快速重新获得全部活性的热启动聚合酶。
当在本文中使用时,术语“dntp”是指脱氧核糖核苷三磷酸。这样的dntp的非限制性实例是datp、dgtp、dctp、dttp、dutp,其也可以以标记的衍生物的形式存在,例如包含荧光标记物、放射活性标记物、生物素标记物。还涵盖具有修饰的核苷酸碱基的dntp,其中所述核苷酸碱基是例如次黄嘌呤、黄嘌呤、7-甲基鸟嘌呤、次黄苷、黄嘌呤核苷、7-甲基鸟苷、5,6-二氢尿嘧啶、5-甲基胞嘧啶、假尿苷、二氢尿苷、5-甲基胞苷。此外,上述分子的ddntp也涵盖在本发明中。
当在本文中使用前,术语“ntp”是指核糖核苷三磷酸。这样的ntp的非限制性实例是atp、gtp、ctp、ttp、utp,其也可以以标记的衍生物的形式存在,例如包含荧光标记物、放射活性标记物、生物素标记物。
本发明还涉及本发明的试剂盒或核酸引物在测序或扩增和检测核酸的方法中的使用。上文中描述的核酸引物的实施方式,也适用于所使用的核酸引物的实施方式。
附图说明
图1:通过内部标记的引物改良基线。使用各0.4μm正向和反向引物,进行不使用模板dna的pcr。随后通过毛细管电泳分析pcr探针。在上方的电泳图中,示出了使用标准的标记引物的无模板对照(ntc)pcr的结果(a)。使用内部标记的引物的ntc-pcr的结果示出在b中。内部标记的引物在70至550bp之间的范围内不引起任何假阳性信号,基线绝对平坦。
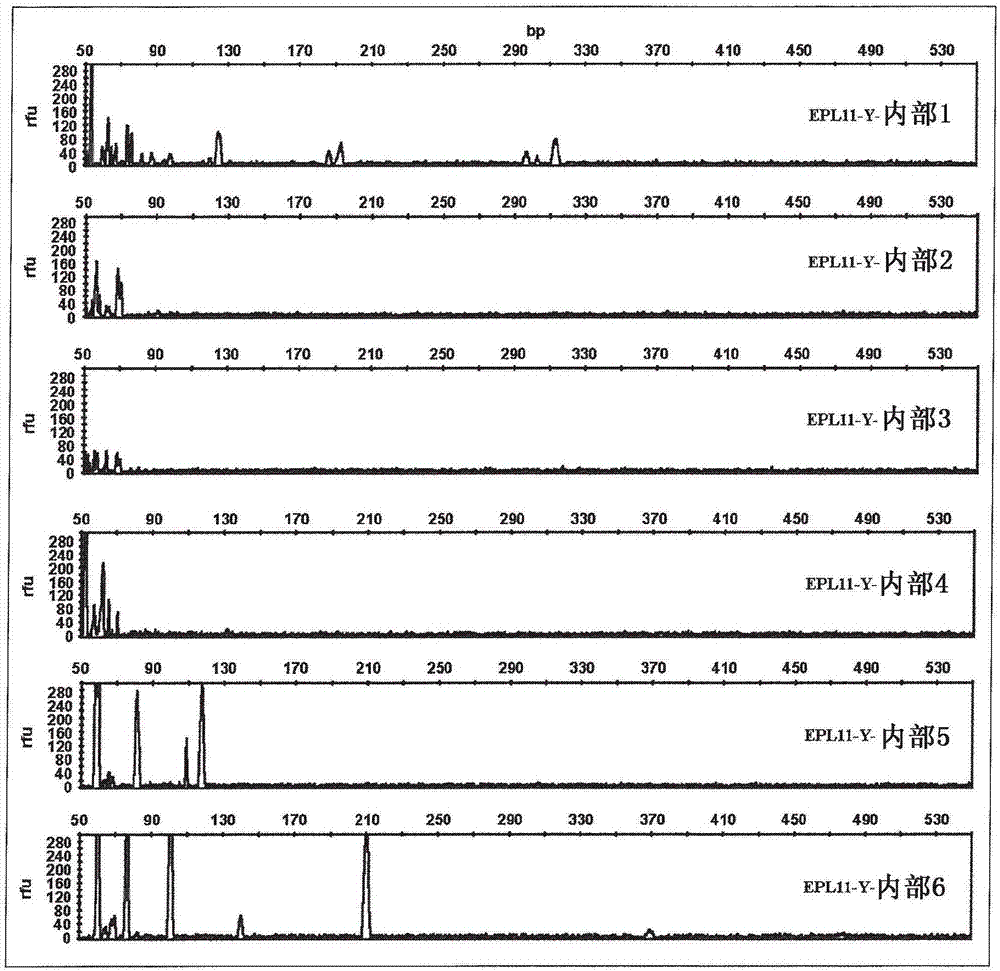
图2:内部染料偶联位置的影响。对引物epl11-y在各种不同内部引物位置处进行荧光标记(参见表1)。示出了使用0.4μm引物(终浓度)但是不使用模板dna的pcr后毛细管电泳的结果。染料越偶联于引物的中间,出现的人为峰越少。
具体实施方式
实施例
实施例1
在这些实施例中的pcr如下进行:
使用0.2μlmultitaq2dna聚合酶(qiagen,hilden,germany)作为热启动dna聚合酶。使用2.5μlreactionmixb(qiagen,hilden,germany)作为pcr缓冲溶液。使用0.4μm内部标记的正向引物d16-p-intern(5’-gggggtctaagagcxtgtaaaaag-3’;seqidno.1;其中x表示连接到胸苷核苷酸的环外氨基的atto550)或0.4μm标记的正向引物d16-p-atto550(5’-atto550-gggggtctaagagcttgtaaaaag-3’;seqidno.2)与0.4μm未标记的d16_rev反向引物(5’-gtttgtgtgtgcatctgtaagcatgtatc-3’;seqidno.3)的组合进行pcr,所有引物由biomers.net(ulm,germany)生产。
表1:
用无核酸酶的水(qiagen,hilden,germany)补足25μl的体积。
pcr使用9700pcrsystemgold热循环仪(lifetechnologiescorporation,carlsbad,ca,usa),按照下列方案来进行:
表2:用于实施例1的循环条件
向12μlhi-diformamid(lifetechnologiescorporation,carlsbad,ca,usa)和0.5μldna尺寸标准品550(bto)(qiagen,hilden,germany)加入1μl循环的pcr探针,并在95℃温育3分钟。
通过毛细管电泳,使用3500geneticanalyzer(lifetechnologiescorporation,carlsbad,ca,usa),在标准条件下分析变性的pcr探针。
在图1中示出的结果清楚地证实,使用内部标记的引物导致普通背景噪音的不想要的人为峰的降低。在70至550bp范围内的基线绝对干净,即没有任何背景峰或染料污迹。相反,在5’末端标记的标准pcr引物产生大量人为峰,其具有高达500个相对荧光单位(rfu)。使用最少量模板dna的pcr扩增通常产生的pcr产物给出200rfu左右的峰。使用标准的标记引物,电泳图的解释受到大量高的人为峰(染料污迹)的损害,导致对探针的不完整的评价。使用内部标记的引物,由于没有可见的染料污迹,可以清楚地辨别和指派200rfu的pcr产物。这些改进在方法的灵敏度方面提供了巨大优势。
实施例2
当使用在任何内部碱基处标记的引物时,背景噪音的强烈减少是明显的。然而,在进一步的实验中显示,通过选择内部标记物在引物中的位置,可以进一步增强这种效果。
引物在epl-11-y序列的不同碱基对位置处被标记(seqidno.4至9;参见表1)。如实施例1中所述进行分析,使用引物epl11/2(5’-gaacaactctggttgtattgtcttc-3’;seqidno.10)作为反向引物和下列循环条件。
表3:用于实施例2的循环条件
图2中示出的结果证实,通过将标记物的内部位置选择成接近引物的中间位置,可以进一步增强基线平滑效果。
序列表
<110>凯杰有限公司
<120>包含内部标记引物的核酸的测序、扩增和检测方法
<130>b051-0080wo1
<150>ep11165142.8
<151>2011-05-06
<160>10
<170>patentinversion3.5
<210>1
<211>24
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwardprimer
<220>
<221>misc_binding
<222>(15)..(15)
<223>atto550linkedtotheexocyclicaminogroupofthymidine
nucleotide
<400>1
gggggtctaagagcttgtaaaaag24
<210>2
<211>24
<212>dna
<213>artificialsequence
<220>
<223>labelledforwardprimer
<400>2
gggggtctaagagcttgtaaaaag24
<210>3
<211>29
<212>dna
<213>artificialsequence
<220>
<223>unlabelledreverseprimer
<400>3
gtttgtgtgtgcatctgtaagcatgtatc29
<210>4
<211>25
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwardprimer(epl-11-y-intern1)
<220>
<221>misc_binding
<222>(3)..(3)
<223>atto550linkedtotheexocyclicaminogroupofathymidine
nucleotide
<400>4
aatgaattgaacaaatgagtgagtg25
<210>5
<211>25
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwardprimer(epl-11-y-intern2)
<220>
<221>misc_binding
<222>(7)..(7)
<223>atto550linkedtotheexocyclicaminogroupofthymidine
nucleotide
<400>5
aatgaattgaacaaatgagtgagtg25
<210>6
<211>25
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwaredprimer(epl-11-y-intern3)
<220>
<221>misc_binding
<222>(8)..(8)
<223>atto550linkedtotheexocyclicaminogroupofathymidine
nucleotide
<400>6
aatgaattgaacaaatgagtgagtg25
<210>7
<211>25
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwardprimer(epl-11-y-intern4)
<400>7
aatgaattgaacaaatgagtgagtg25
<210>8
<211>25
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwardprimer(epl-11-y-intern5)
<220>
<221>misc_binding
<222>(20)..(20)
<223>atto550linkedtotheexocyclicaminogroupofathymidine
nucleotide
<400>8
aatgaattgaacaaatgagtgagtg25
<210>9
<211>25
<212>dna
<213>artificialsequence
<220>
<223>internallylabelledforwardprimer(epl-11-y-intern6)
<220>
<221>misc_binding
<222>(24)..(24)
<223>atto550linkedtotheexocyclicaminogroupofathymidine
nucleotide
<400>9
aatgaattgaacaaatgagtgagtg25
<210>10
<211>25
<212>dna
<213>artificialsequence
<220>
<223>unlabelledreverseprimer(epl11/2)
<400>10
gaacaactctggttgtattgtcttc25
- 还没有人留言评论。精彩留言会获得点赞!