一种芳香氧化偶氮苯化合物及其制备方法与流程
本发明属于有机合成领域,具体涉及一种芳香氧化偶氮苯化合物及其制备方法。
背景技术:
芳香氧化偶氮苯化合物不仅是重要的功能材料,在染料、药物、食品添加剂以及液晶材料等方面有着重要的应用,其还是重要的有机合成中间体,可以进一步还原成芳香偶氮化合物或者氧化成芳香胺;同时,它也可以重排成羟基偶氮苯,为羟基偶氮苯的合成提供了便捷的途径。由于芳香族氧化偶氮化合物与芳香偶氮化合物有着相似的结构,使得其在性质和功能方面与芳香偶氮化合物有着许多共性,因此对芳香氧化偶氮苯化合物性质与功能的研究将是一个重要的课题。基于芳香氧化偶氮苯化合物重要的应用价值,其合成方法已有很多的报道。
然而,在现有合成方法中,或多或少都存在一些不足。传统的合成方法大都使用了有毒的物质或者反应条件较苛刻。葡萄糖还原的方法是最经典的方法,但是它需要较高的反应温度以及严格的投料比,且产率相对较低;并且现有催化剂制备复杂,金属的引入会造成环境污染。因此,需要开发一种高效、低毒、环保的制备芳香氧化偶氮苯化合物的方法。
技术实现要素:
本发明的目的是提供一种芳香氧化偶氮苯化合物的制备方法,在光照下,将芳香硝基化合物还原成芳香氧化偶氮苯化合物,制备简单、收率高、绿色环保。
为达到上述目的,本发明的总体构思是单硝基芳香化合物,在氙灯辐照、氩气氛围条件下,对各底物的硝基进行还原偶联得到芳香族氧化偶氮苯。
本发明的具体的技术方案是:一种芳香氧化偶氮苯化合物,其化学结构式是以下结构式中的一种:
上述芳香氧化偶氮苯化合物的制备方法,主要包括以下步骤:
(1)采用以下制备方法中的一种制备芳香硝基底物;
①以浓硝酸、芴为原料制备得到浅黄色固体;在碱存在下,以浅黄色固体、1-溴辛烷为原料制备得到芳香硝基底物;
②在碱存在下,以咔唑、1-溴十二烷为原料制备得到白色固体;然后以浓硝酸、白色固体为原料制备得到芳香硝基底物;
③以浓硝酸、2-溴芴为原料制备得到浅黄色固体;在碱存在下,以浅黄色固体、1-溴辛烷为原料制备得到芳香硝基底物;
④以茚酮为原料,在乙酸和浓盐酸中,制备得到淡黄色固体;以淡黄色固体、正丁基锂、1-溴己烷为原料制备得到黄色固体;以黄色固体、浓硝酸为原料制备得到芳香硝基底物;
⑤在氧化剂下,以对硝基苯胺、联硼酸频哪醇酯、亚硝酸叔丁酯为原料,制备得到淡黄色固体;在碳酸盐、有机贵金属化合物存在下,以淡黄色固体、9,9二辛烷二溴芴为原料制备得到芳香硝基底物;
⑥在碳酸盐存在下,以4-羟基苯硼酸频哪醇酯、1,6-二溴己烷为原料,制备得到第一化合物;在碳酸盐、有机贵金属化合物存在下,以第一化合物、2-溴-9,9-二辛基-9-氢芴为原料,制备得到第二化合物;在碳酸盐存在下,以第二化合物、4-硝基苯酚为原料制备得到芳香硝基底物;
(2)将芳香硝基底物、甲苯、异丙醇、氢氧化钾混合后在惰性氛围中,氙灯辐照下,反应得到芳香氧化偶氮苯化合物。
本发明所述芳香硝基底物为化合物a、化合物b、化合物c、化合物d、化合物e或者化合物f;
所述化合物a的结构式为:
所述化合物b的结构式为:
所述化合物c的结构式为:
所述化合物d的结构式为:
所述化合物e的结构式为:
所述化合物f的结构式为:
进一步的制备方法,主要包括以下步骤:
(1)采用以下制备方法中的一种制备芳香硝基底物;
①混合有机溶剂与浓硝酸,于0~10℃下,加入芴,反应15~30min后,重结晶、抽滤后得到浅黄色固体;然后将金属碱加入有机溶剂中,再加入浅黄色固体;然后滴加1-溴辛烷,反应25~40min,得到芳香硝基底物;
②冰水浴下将碱加入有机溶剂中,然后加入咔唑;然后滴加1-溴十二烷;然后室温下反应11~15h,得到白色固体;混合有机溶剂与浓硝酸,于0~10℃下,加入白色固体,反应15~30min,然后萃取、重结晶得到芳香硝基底物;
③混合有机溶剂与浓硝酸,于0~10℃下,加入芴,反应15~30min后,重结晶、抽滤后得到浅黄色固体;然后将金属碱加入有机溶剂中,再加入浅黄色固体;然后滴加1-溴辛烷,反应10~15h,得到芳香硝基底物;
④将茚酮加入乙酸和浓盐酸中,100℃下反应15~18h,得到淡黄色固体;取淡黄色固体加入有机溶剂中,惰性气氛保护下,滴加正丁基锂;然后滴加溴己烷,随后反应10~15h,得到黄色固体;冰水浴下,取黄色固体加入有机溶剂中,然后滴加浓硝酸,反应55~70min,得到芳香硝基底物;
⑤在惰性气氛保护下,将对硝基苯胺、联硼酸频哪醇酯以及氧化剂加入有机溶剂中,于20~25℃下,滴加亚硝酸叔丁酯有机溶剂溶液,反应3~5h,得到淡黄色固体;依次将9,9二辛烷二溴芴、碳酸盐溶液、有机贵金属化合物以及有机溶剂加入淡黄色固体中,在80℃下反应10~15h,得到芳香硝基底物;
⑥反应器中依次加入碳酸盐、4-羟基苯硼酸频哪醇酯、有机溶剂,于60℃下,加入1,6-二溴己烷,反应3~5h;得到第一化合物;在惰性气氛氛围中,在反应器中依次加入第一化合物、2-溴-9,9-二辛基-9-氢芴、有机贵金属化合物、有机溶剂、碳酸盐,在80℃下反应6~8h;得到第二化合物;在反应器中依次加入第二化合物、碳酸盐、有机溶剂,在80℃下搅拌15~30min,随后滴加4-硝基苯酚,继续反应5~7h,得到芳香硝基底物;
(2)反应器中依次加入碱、芳香硝基底物、甲苯、异丙醇,所述甲苯和异丙醇体积比为1:1,芳香硝基底物与碱摩尔比为1:2;然后在惰性气氛氛围中,氙灯(功率为250-1000w)/太阳光辐照下,进行反应得到芳香氧化偶氮苯化合物;反应时间为8~20h。
上述技术方案中,在步骤(1)和步骤(2)中,对产物进行提纯处理,提纯步骤为,先用二氯甲烷萃取(20ml*3),然后用无水硫酸钠干燥,抽滤,旋干;旋干后,进行柱层析,展开剂为二氯甲烷和石油醚(pe:dcm=20:1-5:1,体积比),收集所需组分,旋干,真空烘箱干燥,得到产物。
上述技术方案中,浓硝酸质量浓度为65%;碱为koh;有机溶剂为二氯乙烷、二甲基亚砜、丙酮、四氢呋喃、乙腈、n,n-二甲基甲酰胺;浓盐酸的质量浓度为35%;惰性气氛为氩气气氛;氧化剂为过氧化二苯甲酰;碳酸盐为碳酸钾;有机贵金属化合物为四(三苯基膦)钯。
上述技术方案中,反应体系组分少,条件温和,产率高,选择性高,反应过程中无偶氮苯及芳香胺生成。
光作为一种清洁能源,在有机合成领域有着重要的应用,但是传统的方案中都需与过渡金属或染料结合,从而对环境造成污染以及资源浪费。本发明首次直接使用氙灯辐照芳香硝基底物制备芳香氧化偶氮苯化合物,而在反应过程中不添加任何过渡金属或染料等物质,更加的环保高效。
本发明还公开了上述芳香硝基底物及其制备方法,以及在碱存在下制备芳香氧化偶氮苯化合物中的应用。
由于上述方案的实施,本发明与现有技术相比,具有以下优点:
1.本发明公开制备芳香氧化偶氮苯化合物过程中,条件温和,组分简单,产率高,副反应少,选择性高,反应过程中无芳香偶氮化合物以及芳香胺生成。
2.本发明中利用氙灯辐照芳香硝基底物合成芳香氧化偶氮苯化合物,与现有的合成方法相比,成本低,操作简单,不使用金属离子,低毒环保,更加符合绿色工业的发展要求。同时在太阳光的照射下,也可高效地反应。
附图说明
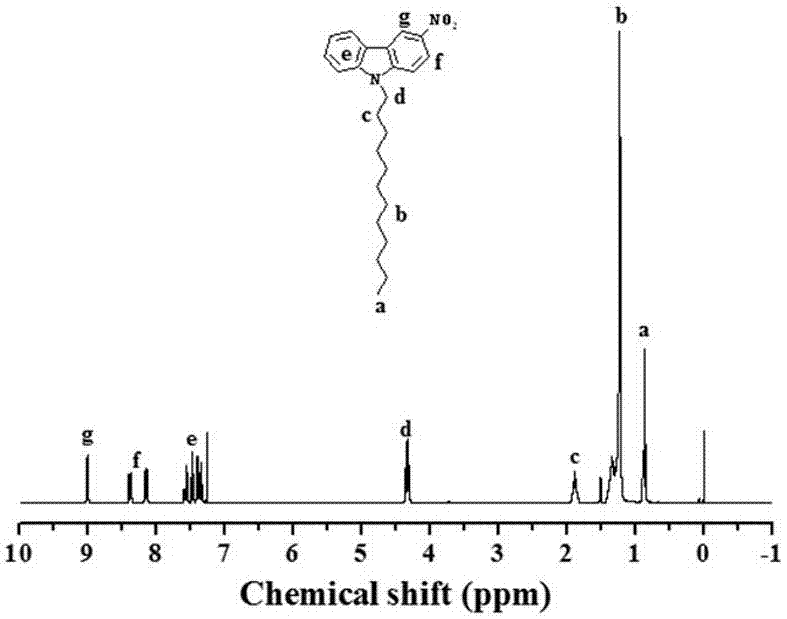
图1为化合物a的核磁图;
图2为化合物b的核磁图;
图3为化合物c的核磁图;
图4为化合物d的核磁图;
图5为化合物e的核磁图;
图6为化合物f的核磁图;
图7为化合物a制备的芳香氧化偶氮苯化合物的核磁图;
图8为化合物b制备的芳香氧化偶氮苯化合物的核磁图;
图9为化合物c制备的芳香氧化偶氮苯化合物的核磁图;
图10为化合物d制备的芳香氧化偶氮苯化合物的核磁图;
图11为化合物e制备的芳香氧化偶氮苯化合物的核磁图;
图12为化合物f制备的芳香氧化偶氮苯化合物的核磁图。
具体实施方式
下面结合实施例和附图对本发明作进一步描述:
本实施例中所用化学试剂:芴,99%,aldrich;2-溴芴,99%,aldrich;四(三苯基膦)钯,99%,aldrich;联硼酸频哪醇酯,99%,aldrich;对硝基苯胺,98%,tci;对羟基苯硼酸频哪醇酯,98%,adamas;茚酮,98%,aldrich;1-溴己烷,99%,中国医药(集团)上海化学试剂公司;1,6-二溴己烷,98%,中国医药(集团)上海化学试剂公司;二甲亚砜,99%,中国医药(集团)上海化学试剂公司;咔唑,97%,中国医药(集团)上海化学试剂公司;无水硫酸钠、氢氧化钾、异丙醇、甲苯、乙腈、乙酸、盐酸、硝酸,分析纯,中国医药(集团)上海化学试剂公司;四氢呋喃(thf)、二氯甲烷、乙醇、正己烷和乙酸乙酯,分析纯,江苏强盛功能化学股份有限公司。
测试仪器及条件:核磁共振仪:300兆赫;测定条件:以cdcl3为溶剂,以四甲基硅烷为内标物,测试温度为室温。
实施例一芳香硝基底物的合成
化合物a的合成:
在250ml烧瓶中加入80ml1,2二氯乙烷,然后加入20ml浓硝酸(65%),使其分散均匀,在冰水浴中反应,随后缓慢加入10g芴,反应20min后,将其倒入到300ml冰甲醇中,抽滤后得到浅黄色固体2(11g)。将7.5gkoh研成粉末加入到100mldmso中,分散均匀,然后缓慢加入上述浅黄色固体6.3g,搅拌20min,随后将15ml1-溴辛烷滴加到上述体系中,反应30min,用石油醚萃取,干燥,旋干,得到粗产物,然后用石油醚作为展开剂进行柱层析得到黄色粘稠液体,即化合物a(9.0g)。
化合物b的合成:
在250ml烧瓶中加入150ml丙酮,冰水浴下将5.4g氢氧化钾研磨成粉末加入到烧瓶中,搅拌10min,随后将5g咔唑缓慢的加入到上述体系中,搅拌30min,接着将11.2g1-溴十二烷滴加到上述体系中,滴完后,室温下反应12h,反应结束后,用二氯甲烷萃取,随后用石油醚作为展开剂进行柱层析得到白色固体4(7.2g);将0.4ml硝酸(65%)加入到100ml1,2二氯乙烷中,控制温度在0-10℃之间,搅拌均匀后,将2g上述白色固体加入到烧瓶中,反应20min,反应结束后,用二氯甲烷萃取,随后用乙醇重结晶得到黄色固体,即化合物b(1.8g)。
化合物c的合成:
在250ml圆底烧瓶中加入80ml1,2二氯乙烷,然后加入20ml浓硝酸(65%),使其分散均匀,随后室温下缓慢加入10g2-溴芴,反应20min后,将其倒入到300ml冰甲醇中,抽滤后得到浅黄色固体6(10g)。将10gkoh研成粉末加入到80mldmso中,分散均匀,然后缓慢加入上述浅黄色固体5g,搅拌20min,随后将7.3g1-1-溴辛烷滴加到上述体系中,反应12h,用乙酸乙酯萃取,无水硫酸钠干燥,旋干,得到粗产物,然后用石油醚作为展开剂进行柱层析得到淡黄色固体,即化合物c(7.5g)。
化合物d的合成:
将6.8g茚酮加入到30ml乙酸和15ml浓盐酸(35%)中,100℃下反应16h,反应结束后,将其倒入在冰水中,随后抽滤,用大量水洗,再用丙酮和二氯甲烷洗,得到淡黄色固体8(4.5g);取3.18g上述淡黄色固体,加入到50ml四氢呋喃中,氩气保护下,将22ml正丁基锂缓慢滴加到上述体系中,反应30min后,将5ml溴己烷滴加到上述反应瓶中,然后反应4h,随后再滴加22ml正丁基锂,30min后,再滴加入5ml1-溴己烷,随后反应12h,反应结束后,加入氯化铵溶液将反应淬灭,随后用石油醚萃取,并用石油醚做展开剂进行柱层析,得到黄色固体9(6.7g);冰水浴下,取1.7g上述黄色固体加入到120ml1,2二氯乙烷中,然后将0.42ml浓硝酸滴加到上述体系中,滴完后,反应1h,反应结束后,将其倒入到冰水中,随后用乙酸乙酯萃取,之后再进行柱层析(石油醚:乙酸乙酯=20:1)得到黄色固体,即化合物d(1.4g)。
化合物e的合成:
在氩气保护下,将1.38g对硝基苯胺、2.54g联硼酸频哪醇酯以及49mg过氧化二苯甲酰加入到60ml乙腈中,控制温度在25℃,随后将1.55g亚硝酸叔丁酯溶解在10ml乙腈中,滴加到上述体系中,反应4h,反应结束后,旋干,以石油醚:乙酸乙酯=20:1作为展开剂进行柱层析,得到淡黄色固体11(1.4g);取1g上述淡黄色固体加入到25ml舒伦克瓶中,然后依次加入2.24g9,9二辛烷二溴芴、6ml碳酸钾溶液(2mol/l)、20mg四(三苯基膦)钯以及9ml四氢呋喃,随后用双排管冷冻-抽气-充气三次,在80℃下反应12h,反应结束后,用二氯甲烷萃取,随后以纯石油醚作展开剂进行柱层析得到黄色固体,即化合物e(1.95g)。
化合物f的合成:
在250ml圆底烧瓶中依次加入k2co3(2.76g,0.02mol),化合物12(4.4g,0.02mol)以及dmf(60ml),搅拌20min,随后在60℃将1,6-二溴己烷加入到上述体系中,反应4h。反应结束后用二氯甲烷萃取,有机层用无水硫酸钠干燥,旋干后柱层析提纯(展开剂:乙酸乙酯/石油醚=1/50),最终得到纯净的产物13(5.6g)。
在氩气氛围中,在50ml圆底烧瓶中依次加入化合物13(1g,2.6mmol),2-溴-9,9-二辛基-9-氢芴(1.35g,3.1mmol),(ph3)4pd(0)(10mg,0.009mmol),thf(7ml)以及2mk2co3(4ml)。随后在80℃下反应7h。反应结束后用二氯甲烷萃取(30ml*3),有机层用无水硫酸钠干燥,旋干,随后用柱层析提纯(展开剂:乙酸乙酯/石油醚=1/50),最后得到纯净的化合物14(1.36g)。
在50ml圆底烧瓶中依次加入4-硝基苯酚(0.27g,1.97mmol),k2co3(0.27g,1.97mmol)以及dmf(30ml),在80℃下搅拌20min,随后将化合物14(1g,1.64mmol)加入到上述体系中,滴加完后继续反应6h;反应结束后用乙酸乙酯萃取,有机层用无水硫酸钠干燥,旋干,随后用柱层析提纯(展开剂:乙酸乙酯/石油醚=1/10),最后得到纯净的化合物f(0.9g)。
附图1-6分别为化合物a、化合物b、化合物c、化合物d、化合物e、化合物f的核磁谱图。得到的对应产物的产率分别为:70%,82%,84%,78%,95%,80%。
上述制备芳香硝基底物的流程示意如下:
实施例二芳香氧化偶氮苯化合物的制备
在10ml安瓿瓶中依次加入11.2mg氢氧化钾、0.1mmol芳香硝基底物(化合物a、化合物b、化合物c、化合物d、化合物e或者化合物f)、3ml甲苯以及3ml异丙醇,加完后,通氩气10min除氧并封管,随后在氙灯(功率为250-1000w可选)辐照下反应10h,反应结束后,用二氯甲烷萃取,无水硫酸钠干燥,旋干,随后进行柱层析提纯(展开剂:石油醚/二氯甲烷),得到对应的芳香氧化偶氮苯化合物。
附图7-12分别为化合物a、化合物b、化合物c、化合物d、化合物e、化合物f制备的芳香氧化偶氮苯化合物的核磁谱图,得到的对应产物的产率分别为:97%,85%,90%,91%,81%,30%。一般认为共轭性对光还原有关键影响,非共轭化合物很难发生光还原反应,本发明的方法针对几乎不共轭的化合物,依然可以取得30%的收率,克服了现有技术偏见。
实施例三芳香氧化偶氮苯化合物的制备(太阳光光照下)
在10ml安瓿瓶中依次加入11.2mg氢氧化钾、0.1mmol化合物a、3ml甲苯以及3ml异丙醇,加完后,通氩气10min除氧并封管,随后在太阳光下室温反应20h;反应结束后,用二氯甲烷萃取,无水硫酸钠干燥,旋干,随后进行柱层析提纯(展开剂:石油醚/二氯甲烷=20/1-8/1),得到对应的芳香氧化偶氮苯化合物,产率为:93%。
上述制备芳香硝基底物的流程示意如下:
- 还没有人留言评论。精彩留言会获得点赞!