用于肺癌诊断的基因群和试剂盒及诊断方法与流程
本发明涉及基因工程和生物技术检测领域,具体涉及用于肺癌诊断的基因群和试剂盒及诊断方法。
背景技术:
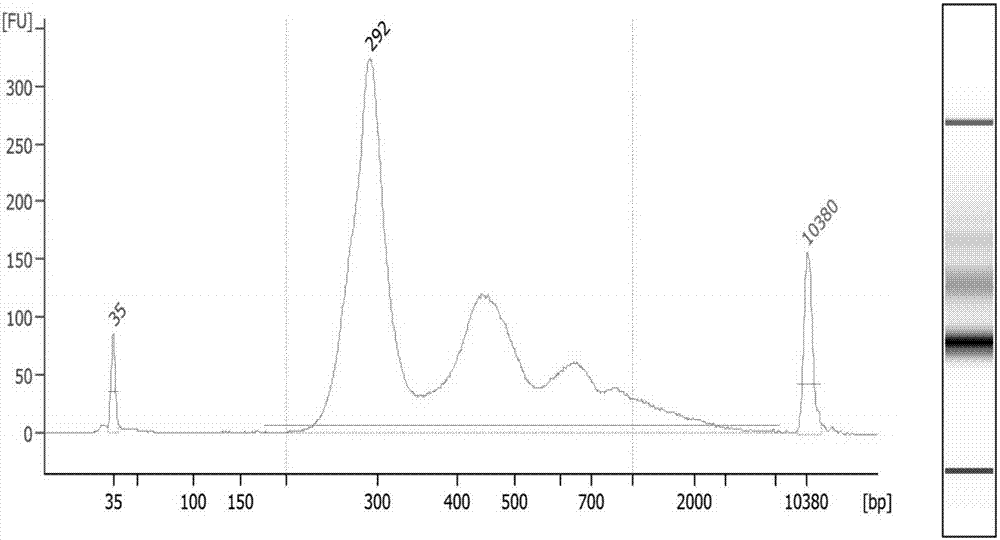
:循环dna是一种无细胞状态的胞外dna,存在于血液、滑膜液和脑脊液等体液中,其主要是由单链或双链dna以及单链与双链dna的混合物组成,以dna蛋白质复合物或游离dna两种形式存在。循环dna作为一种新的肿瘤标志物,将在肿瘤的诊断、治疗及预后检测等方面发挥重要作用,尤其对于一些不具有典型临床症状、检查无特异性和诊断困难的肿瘤可避免复杂的、具有创伤性的活检。循环肿瘤dna(ctdna),是指肿瘤细胞体细胞dna经脱落或者当细胞凋亡后释放进入循环系统,随着基因测序的飞速发展,目前,已能在血液中对其进行检测并计数。ctdna来自肿瘤细胞的体细胞突变,不同于遗传突变的是其存在于体内每个细胞。因此,ctdna是一种特征性的肿瘤生物标记物,并且还可以被定性、定量和追踪。与组织活检相比ctdna检查微创小、无放射性污染、经济、新dna无防腐剂污染等优点,并允许对治疗反应实时监测。针对ctdna的检测有数字pcr、beaming技术、标记扩增深度测序(tam-seq)、癌症个体化深度测序(capp-seq),但这些方法是对已知突变的检测,已知突变的信息获得麻烦,并且检测范围有限。除此外还有全基因组测序、全外显子测序,这些方法不需要已知突变信息,但是成本昂贵。技术实现要素:本发明所解决的现有技术的问题是:针对现有的肺癌检测方法通常存在如下缺陷和不足:第一,传统的检测方法,包括外科手术治疗、放疗和化疗等,不能在肿瘤早期及早发现相关症状;第二,现有的基于肺癌基因的检测方法,通常要求比较高的建库起始量,测序灵敏度和准确性都比较低;第三,已有的用于肿瘤基因检测的文库,其涵盖的基因要么过多,要么过少,过多就是增加了肺癌检测的负担;过少,针对肿瘤相关基因的捕获非常少,会遗漏掉很多重要的基因突变信息;或者现有的检测手段,涵盖有一些与肿瘤不是很相关的基因,不能全面有效的反映肺癌及肺癌相关基因的突变。本发明根据最新前沿肺癌基因组及药物数据,包括cga、美国国家综合癌症网(nccn)、美国食品药品监督管理局(fda)等的肺癌基因组及药物数据筛选入组基因68个。其中包含nccn推荐检测基因、fda已批准靶药和重要临床期药物相应靶基因、影响靶药用药效果的基因、影响化药敏感性及毒副作用的基因等。本发明通过探针捕获技术一次性富集外周血ctdna的68个肺癌基因的外显子区域,实现多基因多靶点并行深度高通量测序,可同时检测基因的多种变异类型(如点突变,缺失,插入等),在检测的深度和准确度上均优于现有的技术。具体而言,本发明提供了用于肺癌诊断的基因群和试剂盒及诊断方法。本发明提供了用于肺癌诊断的基因群,包括以下基因:abcb1,akt1,alk,apc,atic,atm,braf,brca1,brca2,c8orf34,cbr3,cd274,cda,cdk4,cdk6,cdkn2a,ctnnb1,ddr2,dpyd,egfr,erbb2,erbb4,ercc1,fbxw7,fgfr1,fgfr2,fgfr3,flt3,gstp1,hras,jak1,jak2,kdr,kit,kras,map2k1,met,mlh1,mthfr,mtor,mtrr,notch1,nras,ntrk1,ntrk2,pdcd1,pdgfra,pik3ca,ptch1,pten,raf1,rb1,ret,ros1,rrm1,smad4,smo,sod2,stk11,tp53,tpmt,tsc1,ugt1a1,ugt1a9,umps,vegfa,xpc和xrcc1。优选的,根据以上所述的基因群,所述基因群中不同基因包含有外显子和部分内含子,如图9所示。本发明还提供了用于以上所述的基因群诊断的试剂盒。同时,本发明还提供了利用以上所述的试剂盒用于肺癌诊断的方法,包括如下步骤:(1)提取待测样本血浆中游离dna(cfdna)和基因组dna;(2)将基因组dna打断成长度为150~250bp的片段,打断后的基因组dna和游离dna进行杂交捕获,构建dna文库,上机测序,对测序结果分析。优选的,根据以上所述的方法,步骤(2)中样本dna文库的构建包括如下步骤:a.对游离dna样本进行末端修复;b.步骤a得到的连接产物连接a尾;c.将经过a尾连接的样本dna产物与连接接头进行连接。优选的,根据以上所述的方法,还包括如下步骤:在连接a尾后,还包括对a尾连接产物进行纯化的步骤。优选的,根据以上所述的方法,其中所述对a尾连接产物进行纯化是基于固相载体可逆化固定的方法。优选的,根据以上所述的方法,其中,在样本dna连接接头后,还包括对接头连接产物进行纯化的步骤。优选的,根据以上所述的方法,其中,对接头连接产物进行纯化是利用固相载体可逆化固定的方法。优选的,根据以上所述的方法,还包括如下步骤:在步骤a对游离dna样本进行末端修复后,利用磁珠对末端修复的dna样本进行纯化。本发明所取得的有益效果为:本发明通过探针捕获技术一次性富集循环肿瘤dna(ctdna)68个基因的外显子区域,实现多基因多靶点并行深度高通量测序,可同时检测基因的多种变异类型(如点突变,缺失,插入等),优化了检测流程并提高了检测精度,使液体活检、低频检测和肿瘤早诊成为可能。用于捕获68个基因靶区域并行深度高通量测序的技术,且同时用于检查其他基因的多种变异类型,如位点突变,小的缺失和插入等,为实现液体活检提供了一种新的技术策略。附图说明图1为本发明的工作流程图。图2a-2d为以本发明实施例一的方法制备的四样本混合文库的qc检测示意图,通过agilent2100片段分析仪检测使用本发明构建的文库,结果显示成功捕获到了血浆游离的dna,加上接头290bp左右。图3a-3d为以本发明实施例一的方法制备得到的混合文库的测序深度示意图,本发明四个样本的序列测序深度的分布均在15000层左右,没有长拖尾,覆盖均一。图4为插入片段结果示意图,通过对测序数据的分析,该实验的得到的混合文库的插入片段为170bp左右,说明成功捕获到了血浆游离dna。图5为实施例二采用多重pcr扩增cbr3产物的统计结果图。图6为实施例二采用多重pcr扩增cda产物的统计结果图。图7为实施例二采用探针捕获cbr3产物的统计结果图。图8为实施例二采用探针捕获cda产物的统计结果图。图9为本发明68个基因的染色体位置及外显子和部分内含子位置图。具体实施方式以ctdna为检测对象,针对肺癌相关的基因进行突变情况的检测,检测只需要外周血液样品即可,可以真正实现无创检测。本领域技术人员在实际探索过程中也针对肺癌的突变情况进行了多种尝试。申请号为201610363675.1,公开号为cn106047998a的中国专利申请,针对肿瘤相关的105个基因设计12659条捕获探针对构建的cfdna文库进行杂交捕获,然后进行测序,并对测序结果进行数据分析,但是该专利申请中并未记载与肿瘤相关的这105个基因,而且针对105个基因同时检测,工作量大,需要处理的信息大,效率不高;而且该专利申请中检测范围为所声明基因的部分区域或者热点区域,此种方法在预测新发突变尤其是新发基因融合的时候明显受限,不能够有效地把所有突变检测出来,在实际检测结果中出现漏检或者假阴性。而本发明中所涉及到的68个基因为全部基因的外显子和部分重要融合基因的内显子区域(如图9所示),不管涵盖了nccn推荐基因,fda已批准靶药和重要临床期药物相应靶基因、影响靶药用药效果的基因、影响化药敏感性及毒副作用的基因,也针对所有潜在的新发突变进行全面检测,可以为临床诊断和治疗增加有效依据,且不增加检测成本。为此,本发明发明人基于现有的肺癌基因组及药物数据,筛选出68个与肺癌相关的基因,如下表1所示:表1与肺癌相关的68个基因这68个基因的选择范围与依据如下,其中,某些基因同时承担着多个功能,例如tp53既是重要的潜在的药物靶点,又是重要的化疗作用评估对象:a)nccn指南中明确推荐的与非小细胞肺癌靶向用药相关的8个基因(egfr、alk、braf、met、ret、ros1、erbb2、kras)。b)肺癌ⅱ/ⅲ期临床试验在研药物的靶基因、其他癌种已获fda批准药物的靶基因、肺癌高频突变基因,共42个,为患者寻找常规肺癌靶药以外的潜在获益药物(pik3ca、nras、erbb4、smad4、brca2、ptch1、akt1、pdgfra、fbxw7、ntrk1、cdk4、raf1、cdkn2a、pten、fgfr1、apc、cdk6、smo、ctnnb1、rb1、fgfr2、atm、flt3、tsc1、hras、stk11、fgfr3、kdr、jak1、pdcd1、kit、tp53、map2k1、mlh1、jak2、cd274、mtor、ddr2、notch1、brca1、ntrk2和vegfa)。c)常用化疗药物毒副作用评估类基因19个。不同患者对肿瘤化疗药物反应相关基因(如药物转运蛋白基因、药物代谢酶基因、dna修复基因、细胞凋亡相关基因)的snp存在差异,使得患者对特定药物的敏感性或毒副作用反应不同。通过这部分基因的检测对传统广谱化疗药物进行评估,为患者避免选择不良反应过高的化疗药物提供参考,包括abcb1、gstp1、ugt1a1、atic、mthfr、ugt1a9、c8orf34、mtrr、umps、cbr3、rrm1、xpc、cda、sod2、xrcc1、dpyd、tp53、ercc1和tpmt。d)针对肺癌的重要的融合基因,包括alk,ros1和ret。在本发明的一种优选实施方式中,本发明提供了如下制备方法,包括:(1)dna样本的制备:提取不同样本血浆中的游离dna,用于文库构建;(2)dna样本的末端修复:对步骤(1)得到的游离dna进行末端修复;(3)磁珠纯化:利用磁珠对末端修复的dna样本进行纯化,优选利用ampurexp磁珠进行纯化;(4)连接a尾:对纯化产物连接a尾;(5)样本dna连接接头:将经过a尾连接的样本dna产物与连接接头进行连接;(6)对步骤(5)经过接头连接的样本dna进行文库扩增,得到捕获前pcr样本;(7)文库杂交:利用特异性探针同捕获前pcr样本进行杂交,得到ctdna,洗涤获得捕获产物;(8)对捕获产物进行扩增,纯化得到ctdna文库。纳米级磁珠微珠,表面带有一种官能团,能与核酸发生吸附反应,使得核酸与蛋白质或者细胞中的其他物质进行分离,并且可以实现自动化。例如可以利用硅磁,离心磁珠进行吸附,优选利用ampurexp磁珠进行吸附。其中,在一种优选实施方式中,在连接a尾后,还包括对a尾连接产物进行纯化的步骤。以上对a尾连接产物的纯化步骤,优选基于固相载体可逆化固定的方法进行纯化分离。该方法基于以下机理:在一定浓度的peg和盐离子环境中,dna可以吸附到羧基修饰的高分子磁性微球的表面,该吸附过程是可逆的,在适当的条件下,结合的dna分子可以被洗脱回收。其中,在一种优选实施方式中,在样本dna连接接头后,还包括对接头连接产物进行纯化的步骤。以上对接头连接产物进行纯化的步骤,优选基于固相载体可逆化固定的方法进行纯化分离。该方法基于以下机理:在一定浓度的peg和盐离子环境中,dna可以吸附到羧基修饰的高分子磁性微球的表面,该吸附过程是可逆的,在适当的条件下,结合的dna分子可以被洗脱回收。其中,在一种优选实施方式中,在捕获前pcr样本构建成功后,还包括对捕获前pcr产物进行纯化的步骤,优选利用磁珠进行纯化。下面结合各个具体实施方式,对本发明及其有益技术效果进行详细说明,其中本实施例以技术方案为前提,给出了详细的实施方式和具体的操作过程,但本发明的保护范围并不限于本实施例。实施例一按照图1的流程图所示,分别进行样本的提取,文库的构建,上机测序以及数据分析。具体的过程如下:1.游离dna的提取利用ctdna提取试剂盒(货号:154022516,厂家:qiagen公司),可以同时提取4个不同样本血浆中的游离dna(这些样本来自于不同的肿瘤患者),将dna定量后分别溶解在50μlddh2o中,进行文库构建。所需试剂准备如表2所示。表2所需要的试剂试剂名称储存方式准备方式bufferacl*室温室温bufferacw1室温室温bufferacw2室温室温bufferacb室温室温蛋白酶k-20℃冰上bufferave室温室温(1)血浆分离1)将4个采血管垂直放置,让其自然沉淀1-2小时。2)离心机速度调至3000rpm,将4个采血管离心15分钟。3)将离心管离心15分钟后,取一支样品管,旋下管帽,并将其放置在架架上。4)分别将4个离心管中约5ml血浆转移到新的15ml离心管中。将离心机速度调至14000rpm,离心10分钟。5)分别将4个离心管中约5ml血浆转移到新的15ml离心管中,旋紧样品管盖,在样品管壁上做好标记。(2)ctdna提取1)分别吸取500μl的蛋白酶k到4个新的50ml离心管中。2)向50ml离心管中分别加入上述4个样本5ml的血浆。3)向50ml离心管中加入4mlbufferacl(包含10ugcarrierrna)。关上盖子,涡旋震荡30s混匀。4)混匀后立即将离心管置于60℃的水浴中,孵育30min。5)分别将离心管置于离心管架上,小心拧开盖子,分别加入9mlbufferacb于该离心管中,涡旋15-30s后,冰上冰浴5min。6)将上述离心管中的混合液一次通过收集膜管,真空抽滤泵进行抽滤。7)加入750μlbufferacw2到收集膜管中,真空抽滤泵进行抽滤。8)加入750μl无水乙醇到收集膜管中,真空抽滤泵进行抽滤。9)14000rpm空管离心3min后,将收集膜置于一个新的2ml收集管中,打开盖子,置于56℃热块中10min晾干。10)将收集膜置于新的1.5ml离心管中,向膜中加入20-150μl的bufferave,盖上盖子,室温静置3min,14000rpm离心1min收集dna。11)qubit检测提取的5个样本的核酸浓度。2.末端修复利用高通量文库构建试剂盒(kapahtplibrarypreparationkit,货号:kk8000,kapabiosystem)对提取的游离dna进行末端修复,其中,所需要的试剂的储存及准备方式见表3。4个样本dna分别按照表4中所述的体系进行末端修复。表3所需要的试剂储存和制备方式表4末端修复体系pcr级水8μl10×kapaendrepairbuffer7μlkapaendrepairenzymemix5μldna片段(≥10ng)50μl按照表4所述的末端修复体系,将混合液上下吹打混匀后,置于20℃孵育30min。由于每个样本来源个体所含有的血浆游离dna含量不同,提取到的游离dna总量会有不同,但达到10ng便可以进行文库的构建。3.ampurexp磁珠纯化按照(agencourtampurexp,a6388,贝克曼)说明书中所述,利用ampurexp磁珠进行纯化。1)让ampurexpbead在室温放置至少30min,充分混匀ampurexpbead悬浮液;2)在1.5ml新管中加入1.7倍体积混匀的ampurexpbeads悬浮液和修复后的4个dna样品,吹吸混匀10次以上,室温放置5min;3)短暂离心,将离心管放在磁力架上,静置大约3-5min至溶液变澄清;4)在磁力架上小心地吸除管中的上清,枪头不要碰到磁珠;5)在磁力架上,在每个管中分别加入200μl80%乙醇,放置30s,弃乙醇;6)重复步骤5);7)在室温放置5min,使管中残留的乙醇完全蒸发;不能过分蒸干以防止降低产量4.连接a尾和纯化a尾连接产物利用高通量文库构建试剂盒(kapahtplibrarypreparationkit,货号:kk8000,kapabiosystem)进行接头的连接,按照表6所述的a尾连接体系连接a尾,其中,a尾连接试剂储存和准备方式如表5所示。表5a尾连接试剂储存和准备方式将上述磁珠分别溶解在50μla尾连接体系中,体系见表6。表6a尾连接体系pcr级ddh2o42μl10×kapaa-tailingbuffer5μlkapaa-tailingenzyme(10u/ul)3μlxpbeads-dna--将混合液上下吹打混匀后,置于30℃孵育30min。然后按照agencourtampurexp,a6388,贝克曼说明书描述,纯化a尾连接产物。5.样本dna连接接头按照高通量文库构建试剂盒(kapahtplibrarypreparationkit,货号:kk8000,kapabiosystem)说明书和dna接头试剂盒(货号7141530001,seqcapadapterkita96rsn,购自于上海捷易生物科技有限公司)描述,参照表8所述体系对样本dna连接接头。其中,在连接接头时所用到的试剂的储存方式和使用时的准备方式如表7所示。表7dna连接接头所用试剂的储存和准备方式所需试剂储存方式准备方式5×kapaligationbufferkapahtplibpreparationkitillumina-20℃冰上kapat4dnaligasekapahtplibpreparationkitillumina-20℃冰上pcr级水kapahtplibpreparationkitillumina-20℃冰上peg/naclspri溶液kapahtplibpreparationkitillumina-20℃冰上adapterseqcapadapterkita-20℃冰上乙醇室温室温将上述磁珠分别溶解在47μl连接接头的体系中,体系如表8所示。表8连接接头体系pcr级ddh2o32μl5×kapaligationbuffer10μlkapat4dnaligase(13u/ul)5μlxpbeads-dna--将体系混匀后,每个样本中加入3μl含有不同index的接头。上下吹混数次后,置于20℃孵育15min。然后按照agencourtampurexp,a6388,贝克曼说明书描述,纯化接头连接产物。6.捕获前扩增按照高通量文库构建试剂盒(kapahtplibrarypreparationkit,货号:kk8000,kapabiosystem)说明书描述,参照表10所述体系对捕获前ctdna进行扩增。其中,在ctdna捕获前扩增所用到的试剂的储存方式和使用时的准备方式如表9所示。表9ctdna捕获前扩增所用试剂的储存和准备方式扩增前将引物用ddh2o溶解,并将pre-capturelm-pcrmastermix置于冰上溶解,配置如下反应体系:表10ctdna捕获前扩增所用到的反应体系将上述混合液混匀并置于pcr仪中,执行如下程序:98℃预变性45s;98℃变性15s,60℃退火30s,70℃延伸30s,循环10次;72℃后延伸1min,置于4℃。反应结束后得到的即是捕获前pcr样本。其中,引物序列prelm-pcroligos1为seqidno.1,引物序列prelm-pcroligos2为seqidno.2,seqidno.1序列为:aatgatacggcgaccaccgaseqidno.2序列为:caagcagaagacggcatacga。然后按照agencourtampurexp,a6388,贝克曼说明书描述,对扩增后的捕获前ctdna进行纯化。7.文库杂交dna探针是带有生物素标记的寡核苷酸序列,与变性的dna序列结合,特异性识别目的片段。杂交时用seqcapheuniversaloligo和seqcapheindexoligo进行adaptor无用序列的封闭(由罗氏nimblegenseqcaphybridizationandwashkit,货号:05634253001,roche试剂盒中提供),并用cothumandna阻断非特异性杂交。其中cothumandna源于人类胎盘dna,含有大量的重复序列,用于阻断非特异性杂交。杂交结束后用链霉素亲和磁珠结合dna探针上的生物素标记,以结合目的dna片段,随即用相应的洗脱液进行洗脱,扩增,出库,测序。按照nimblegenseqcaphybridizationandwashkit,05634253001,roche说明书描述,参照表12所述体系与捕获前文库进行杂交。其中,文库杂交时所用到的试剂的储存方式和使用时的准备方式如表11所示。表11文库杂交时所用到的试剂的储存方式和使用时的准备方式1)杂交前的准备,分别溶解seqcapheuniversaloligo和seqcapheindexoligo至终浓度为1000um置于-20℃.2)准备杂交混合样本,将4个不同index的样本定量,并且相同比例混合至1.0ug。3)准备提高混合杂交的引物池,在冰上溶解1000um的seqcapheuniversaloligo和seqcapheindexoligo,混合比例为50%的seqcapheuniversaloligo和50%的seqcapheindexoligos,总引物量为2000pmol,具体实施如下表所示。表12组分含量seqcapheuniversaloligo1000pmol(1μlof1000um)seqcapheindex1oligo2000pmol(0.25μlof1000um)seqcapheindex2oligo2000pmol(0.25vμlof1000um)seqcapheindex3oligo2000pmol(0.25μlof1000um)seqcapheindex4oligo2000pmol(0.25μlof1000um)4)准备杂交样本4-1)取5μlcothumandna(1mg/ml)加入1.5ml离心管中4-2)将1.25ug(5μl)的混合文库加入到4-1的离心管中4-3)将2000pmol的混合引物seqcapheuniversaloligo和seqcapheindexoligo各1μl加入到离心管中。将上述混合液置于具孔的离心管中并置于60℃真空浓缩,向管中加入7.5μl2×hybridizationbuffer和3μlofhybridizationcomponenta,充分混匀,以最大转速离心10s后置于95℃孵育10min。以最大转速离心10s。向上述管中加入4.5μl的杂交探针seqcapezprobepool,最终混合物如下表13。表13混合物体系cothumandna(1mg/ml)5ug混合文库1.25ugseqcapheuniversaloligo1000pmolseqcapheindexoligo1000pmol2×hybridizationbuffer7.5μlhybridizationcomponenta3μlseqcapezprobepool4.5μltotal15μl将上述混合物混匀后,置于47℃中放置17h,设置温度为57℃。然后按照seqcap杂交和洗涤试剂盒(seqcaphybridizationandwashkit,罗氏nimblegen)说明书的描述,对于捕获产物进行洗涤回收。8.捕获后文库扩增利用高通量文库构建试剂盒(kapahtplibrarypreparationkit,购自上海希匹吉生物技术有限公司)对捕获后的文库进行扩增,其中所用到的试剂准备如下表14。表14文库扩增的试剂的存储和准备方式扩增前将引物用ddh2o溶解,并将post-lm-pcrmastermix置于冰上溶解,配置如下反应体系,见表15。表15文库扩增的试剂体系kapahifihotstartreadymix(2×)25μlpost-lm-pcroligos1&2(5μm)5μl涡旋文库使磁珠混匀,取20μl加入到上述混合体系中,混匀并置于pcr仪中,执行如下程序:98℃预变性45s;98℃变性15s,60℃退火30s,72℃延伸30s,循环13次;72℃后延伸1min,反应结束后得到扩增后的文库,然后置于4℃保存。按照(agencourtampurexp,a6388,贝克曼)说明书中所述,利用ampurexp磁珠对扩增后的文库进行纯化,制备得到最终的文库。9.文库质量检测,检测通过后即可上机测序,并对测序结果进行数据分析10.原始数据过滤1)去除adaptor污染序列;2)去除含n占整条reads10%以上的序列;3)去除低质量序列,保留q20>=90%的序列;4)去除reads的前端5bp碱基质量低的序列。11.数据比对1)以ucsc中的已知人类的基因组为参考序列(human,grch37,http://hgdownload.cse.ucsc.edu/goldenpath/hg19),使用bwa(burrows-wheeleraligner,https://sourceforge.net/projects/bio-bwa/files/)对过滤后的测序数据进行初步比对;2)使用samtoolsview–bs转换bam文件为sam文件,并使用samtoolssort-@进行重新排序;3)使用picard-toolsmarkduplicates.jar去除数据重复序列(duplicates),降低文库构建过程中可能引入的pcr偏差;4)为降低比对的错误率,将比对到indel附近的reads进行局部重新比对4-1)首先使用gatkrealignertargetcreator确定重新比对的区域;4-2)再在这些区域内使用gatkindelrealigner重新比对。5)对bam文件中的reads的碱基质量值进行重新矫正;5-1)利用gatkbaserecalibrator,已知位点参考文件选取dbsnp_137.hg19.vcf、mills_and_1000g_gold_standard.indels.hg19.vcf、1000g_phase1.indels.hg19.vcf,生成质量值矫正所需文件;5-2)根据质量值矫正文件与矫正前数据进行比较,生成碱基质量值矫正前后比较图;5-3)使用gatkprintreads将经过质量值校正的数据输出到最终的bam文件中。12.变异检测1)使用mutect软件,参考cosmic和dbsnp数据库信息,对检测样本的snv进行初步检验;2)使用gatksomaticindeldetector,对检测样本的indel进行初步检验;3)根据芯片区间信息,对检测样本的比对文件提取所有区间位点的深度数据;4)结合位点的深度数据,对初步检测得到的snv和indel数据进行过滤,将深度低于10x的位点去除;13.变异注释1)根据68个基因的芯片区间范围,对检测到变异位点进行提取;2)使用annovar软件,对提取的snv进行基因变异注释,其中基因组参考hg19,变异位点参考dbsnp;3)使用annovar软件,对提取的indel进行基因变异注释,其中基因组参考hg19,变异位点参考dbsnp;4)参考cosmic数据库,对变异位点进行体细胞变异注释。5)根据积累的突变位点适用的靶向药物信息,对部分变异位点进行靶向用药信息注释。以下将通过对四个样本,按照如上方法进行试验得到混合文库,对该混合文库的实验结果做如下检测,并对本发明的性能进行分析,结果见图2到图4所示。其中,图2a-2d为以本发明方法制备的四样本混合文库的qc检测示意图,通过agilent2100片段分析仪检测使用本发明构建的文库,结果显示成功捕获到了血浆游离的dna,加上接头290bp左右。图3a-3d为以本发明方法制备得到的混合文库的测序深度示意图。本发明的测序深度为目标区间单个碱基被测序的平均次数,数值上等于测序得到的总碱基量与捕获区间基因组大小(芯片大小)的比值。本发明四个样本的序列测序深度的分布均在15000×左右(捕获区间上每个碱基平均被测序了15000次),没有长拖尾,覆盖均一。图4为插入片段结果示意图,通过对测序数据的分析,该实验的得到的混合文库的插入片段为170bp左右,说明成功捕获到了血浆游离dna。从图2到图4的结果可以说明从测试样本文库中提取到有效片段,能够满足上机测序的要求,所得数据符合ctdna片段大小分布。实施例二以cbr3和cda基因为例,分别研究了采用多重pcr的方式和实施例一记载的探针捕获测序的方式对cbr3和cda基因进行,对扩增和捕获样本进行建库测序。采用的样本为肺癌血浆游离dna提取产物,起始cfdna的浓度1.4ng/μl。表16不同基因的不同外显子区域用到的不同的引物序列利用nimblegenseqcaphybridizationandwashkit试剂盒,分别采用多重pcr(30-35circles)的方法将目的片段扩增出来,检测产物浓度并建库测序。同时采用实施例一的高通量测序方法,进行文库构建并捕获测序。结果见图5-图8。其中图5和图6为采用多重pcr扩增cbr3和cda产物的统计图,图7和图8为采用探针捕获cbr3和cda产物的统计图。从这些对比图可以看出,由于多重pcr扩增的不均一性,导致部分目的片段扩增不均一,片段数量不等,测序深度等不符合要求。高通量探针捕获测序在产物数量和均一性方面更为良好。综上,本发明通过探针捕获技术一次性富集循环肿瘤dna(ctdna)68个基因的外显子区域和部分内含子区域,可以实现多基因多靶点并行深度高通量测序,可同时检测基因的多种变异类型(如点突变,缺失,插入等),优化了检测流程并提高了检测精度,使液体活检、低频检测和肿瘤早诊成为可能。而且根据附表a所示的基因的外显子区域和部分内含子区域,可以设计不同的探针序列用于捕获68个基因靶区域。同时应用该技术可以检查其他基因的多种变异类型,如位点突变,小的缺失和插入等,为实现液体活检提供了一种新的技术策略。以上所述,仅是本发明实施的较佳实施例,并非对本发明做任何形式上的限制,凡在本发明的精神和原则之内所做的修改、等同替换和改进等,均需要包含在本发明的保护范围之内。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1