阻燃聚乙烯组合物及其发泡珠粒的制作方法
本发明属于高分子材料技术领域,具体涉及一种阻燃乙烯组合物及其发泡珠粒。
背景技术:
聚乙烯(pe)是目前世界上应用最为广泛、产量及产能最大的合成树脂,年需求增长率5%。作为热塑性塑料中的第一大品种,具有质轻、无毒、耐化学腐蚀性优异等特点,广泛应用于电器、化工、食品、机械等行业。但是聚乙烯在实际应用中存在如下缺点。一方面,聚乙烯的氧指数很低,仅为17.5,属于易燃材料。聚乙烯在燃烧时,会产生严重的熔融滴落行为,这给其阻燃改性带来很大的困难。另一方面,聚乙烯的体积电阻率和表面电阻率都很高,这就使其在加工和使用过程中一旦带上静电就很难消除。以上两个缺点给聚乙烯的使用带来了很大的安全隐患。
解决聚乙烯的阻燃问题的途径是加入阻燃剂,阻燃剂主要分类两类:一种是添加型阻燃剂;另一种是反应型阻燃剂。添加型阻燃剂是指同高分子材料进行物理共混发挥阻燃作用的阻燃剂。反应型阻燃剂是通过将一些含有阻燃元素的单体参与聚合反应,使聚合物的主链或侧链中带有阻燃元素,起到阻燃的效果,因此其阻燃效果持久,而且对材料的物理力学性能影响较小,但是因为技术和价格的问题,使其推广和应用受到了很大限制。与反应型阻燃剂相比,添加型阻燃剂使用方便、价格低廉,因而是目前实现聚乙烯阻燃的常用方法,常用于聚乙烯阻燃改性的添加型阻燃剂有卤系阻燃剂、无卤阻燃剂和复合阻燃体系等。
在聚乙烯的生产、加工和应用过程中,静电产生是非常普遍的。然而,聚乙烯的体积电阻率一般在1016-1020ω·cm的范围内,静电荷产生后很难消除。这些电荷积聚容易导致加工及使用过程中,火灾、爆炸和电击等危险事故的发生。采用的方法主要有两类:一类是添加具有表面活性的抗静电剂,使其中的亲水基团增强表面吸湿性,形成一层单分子层的导电膜,从而加快静电荷的漏泄;另一类是添加具有一定导电性能的添加剂或导电树脂,利用其在塑料共混体系中形成的导电通道起抗静电效果,这种共混物称为复合型导电高分子材料或导电高分子合金。对聚乙烯而言,其体积电阻率降到1012ω·cm,即已达到抗静电的要求。
然而,现有的聚乙烯树脂在制备聚乙烯发泡珠粒时存在阻燃及抗静电性能差,且经阻燃抗静电改性后,聚乙烯发泡珠粒的泡孔形貌及发泡倍率控制出现问题,影响后续模塑成型应用。因此,目前存在的问题是急需研究开发一种兼具良好的阻燃性能和抗静电性能的阻燃聚乙烯组合物及其发泡珠粒。
技术实现要素:
本发明所要解决的技术问题是针对上述现有技术的不足,提供一种阻燃聚乙烯组合物及其发泡珠粒。本发明的发明人经过深入研究后发现,采用具有特定熔融指数和特定密度的组分a、组分b和组分c配合使用而得到的聚乙烯基础树脂制成的发泡珠粒泡孔致密均匀,且制成的发泡珠粒成型体的压缩强度非常高;同时,将本发明提供的聚乙烯基础树脂与特定的阻燃剂与特定的抗静电剂配合使用时,阻燃剂和抗静电剂之间可以发生协同作用,同时提高阻燃性能和抗静电性能。由本发明的阻燃聚乙烯组合物制备的发泡珠粒具有高温抗冲击性能佳、抗静电、阻燃、工艺简便、闭孔率高、密度可控的特点。
为此,本发明第一方面提供了一种阻燃聚乙烯组合物,其包括:聚乙烯基础树脂、阻燃剂以及任选地抗氧剂,其中所述阻燃剂包括氧化膦与过渡金属盐形成的配合物。
根据本发明所述的阻燃聚乙烯组合物,所述聚乙烯基础树脂含有组分a、组分b和组分c;
所述组分a为乙烯/α烯烃共聚的线性低密度聚乙烯,其在温度为190℃、载荷为2.16kg下的熔融指数mia为0.01-2g/10min,优选为0.01-1.5g/10min,更优选为0.01-1g/10min;密度ρa为0.880-0.936g/cm3,优选为0.910-0.930g/cm3,更优选为0.915-0.926g/cm3;
所述组分b为乙烯/α烯烃共聚的线性低密度聚乙烯,其在温度为190℃、载荷为2.16kg下的熔融指数mib为2.1-9.9g/10min,优选为3-8g/10min,更优选为3-5g/10min;密度ρb为0.910-0.930g/cm3,优选为0.913-0.928g/cm3,更优选为0.913-0.924g/cm3;
所述组分c为乙烯/α烯烃共聚的线性低密度聚乙烯,其在温度为190℃、载荷为2.16kg下的熔融指数mic为10-80g/10min,优选为10-60g/10min,更优选为15-40g/10min;密度ρc为0.880-0.930g/cm3,优选为0.905-0.928g/cm3,更优选为0.910-0.926g/cm3。
在本发明中,所述熔融指数均按照gb/t3682-2000中规定的方法进行测定,其中,测试条件包括温度为190℃,载荷为2.16kg。
在本发明的一些优选的实施方式中,所述聚乙烯基础树脂中组分a、组分b和组分c的密度ρa、ρb和ρc之间的关系满足-0.04≤ρa-ρb≤0.02且-0.04≤ρa-ρc≤0.02,这样能够使该聚乙烯基础树脂具有更好的发泡性能,并且由该聚乙烯基础树脂制成的聚乙烯发泡珠粒具有更致密均匀的泡孔结构、制成的聚乙烯发泡珠粒成型体具有更高的压缩强度。
所述组分a、组分b和组分c均为乙烯/α烯烃共聚的线性低密度聚乙烯,其中,线性结构是指分子链中仅含有短支链结构,而不含有长支链结构和交联结构,其由聚合单体和聚合工艺条件所决定,具体为本领域技术人员公知,在此不作赘述。
根据本发明所述的阻燃聚乙烯组合物,为了使得到的聚乙烯基础树脂具有更好的发泡性能,在所述聚乙烯基础树脂中,
所述组分a的重量份数wa为25-90重量份,优选为30-80重量份;
所述组分b的重量份数wb为0.1-10重量份,优选为0.5-8重量份;
所述组分c的重量份数wc为10-75重量份,优选为20-70重量份。
在本发明的一些优选的实施方式中,所述组分a的重量份数wa和组分c的重量份数wc与组分a的熔融指数mia的关系优选满足(5.2×lgmia+11.6)≥wa/wc≥(0.9×lgmia+2.1),更优选满足(2.9×lgmia+6.8)≥wa/wc≥(1.1×lgmia+2.7),这样能够使该聚乙烯基础树脂具有更好的发泡性能,并且由该聚乙烯基础树脂制成的聚乙烯发泡珠粒具有更致密均匀的泡孔结构、制成的聚乙烯发泡珠粒成型体具有更高的压缩强度。
根据本发明所述的阻燃聚乙烯组合物,所述聚乙烯组合物在温度为190℃、载荷为2.16kg下的熔融指数为0.1-20g/10min,优选为0.5-10g/10min。在将具有上述特定熔融指数和密度的组分a、组分b和组分c配合使用的基础上,将所述聚乙烯基础树脂整体的熔融指数控制在上述范围内,能够使得到的阻燃聚乙烯组合物具有非常优异的发泡性能。
根据本发明所述的阻燃聚乙烯组合物,所述组分a和组分b的分子量分布指数均满足mw/mn≤4.5,优选满足2.0≤mw/mn≤4.2。所述组分c的分子量分布指数满足mw/mn≤8.0,优选满足3.5≤mw/mn≤6.0。具体地,为了获得具有上述分子量分布的组分a、组分b和组分c,所述组分a和组分b均采用茂金属催化剂聚合得到,所述组分c采用齐格勒-纳塔催化剂聚合得到。其中,所述茂金属催化剂和齐格勒-纳塔催化剂的种类可以为本领域的常规选择,所述茂金属催化剂通常由茂金属化合物和有机铝化合物以及任选的给电子体组成,具体为本领域技术人员公知,在此不作赘述。本发明的发明人经过深入研究后发现,将采用茂金属催化剂聚合得到的具有上述熔融指数和密度的组分a和组分b以及采用齐格勒-纳塔催化剂聚合得到的具有上述熔融指数和密度的组分c配合使用,得到的聚乙烯基础树脂在利用反应釜浸渍法制备发泡珠粒时具有良好的发泡性能且所得发泡珠粒具有良好的泡孔结构,其模塑成型体还具有非常高的压缩强度,非常适用于家居及汽车材料。
本发明对所述组分a、组分b和组分c中α烯烃共聚单体的含量没有特别地限定,例如,所述组分a、组分b和组分c中α烯烃共聚单体的摩尔含量可以各自独立地为0.2-15mol%,优选为1.5-10mol%。在本发明中,α烯烃共聚单体的摩尔含量是指由α烯烃聚合形成的结构单元的摩尔量占总单体结构单元的摩尔量的比例。此外,所述组分a、组分b和组分c中的α烯烃各自独立地选自c3-c20烯烃中的至少一种。从原料易得性的角度出发,所述组分a、组分b和组分c中的α烯烃优选选自丙烯、1-丁烯、2-丁烯、3-甲基-1-丁烯、4-甲基-1-丁烯、1-戊烯、3-甲基-1-戊烯、4-甲基-1-戊烯、3,3-二甲基-1-戊烯、3,4-二甲基-1-戊烯、4,4-二甲基-1-戊烯、1-己烯、4-甲基-1-己烯、5-甲基-1-己烯、1-庚烯、2-庚烯、1-辛烯、1-癸烯、1-十二碳烯、1-十四碳烯、1-十六碳烯、1-十八碳烯和1-二十碳烯中的至少一种,更优选选自1-丁烯、1-己烯和1-辛烯中的至少一种。
考虑到当将该聚乙烯基础树脂制成聚乙烯发泡珠粒时,水下切粒挤出步骤需要熔体泵提高模头的熔体压力,会间接增加能耗,优选地,所述聚乙烯基础树脂还含有润滑剂,这样能够改善所述聚乙烯基础树脂的挤出加工性能和牵条切粒性能。所述润滑剂的种类和用量均可以为本领域的常规选择,例如,所述润滑剂可以选自聚乙二醇(peg)类润滑剂、含氟聚合物类润滑剂、有机硅类润滑剂、脂肪醇类润滑剂、脂肪酸类润滑剂、脂肪酸酯类润滑剂、硬脂酸酰胺类润滑剂、脂肪酸金属皂类润滑剂、烷烃及氧化烷烃类润滑剂和微纳米粒子类润滑剂中的至少一种。具体地,所述peg类润滑剂例如可以为数均分子量为500-50000的peg分子,其可以经过封端、接枝、交联处理,也可以经过其他化学改性或物理改性。所述含氟聚合物类润滑剂例如可以为聚四氟乙烯、聚偏氟乙烯、聚六氟丙烯等中的至少一种,也可以为其他单峰或多峰的含氟聚合物以及结晶或半结晶的含氟聚合物。所述有机硅润滑剂可以为现有的各种以碳、硅原子为分子主链,以甲基、苯基、烷氧基、乙烯基等有机基团的低聚物或齐聚物为侧链的化合物。所述脂肪醇类润滑剂例如可以为软脂肪醇、硬脂肪醇、牛油脂肪醇等中的至少一种。所述脂肪酸类润滑剂例如可以硬脂酸和/或12-羟基硬脂酸。所述脂肪酸酯类润滑剂例如可以为硬脂酸丁酯、硬脂酸单甘油脂、棕榈酸十六烷基酯、硬脂酸十八烷基酯等中的至少一种。所述硬脂酸酰胺类润滑剂例如可以为硬脂酸酰胺、油酸酰胺、芥酸酰胺、n,n-乙撑双硬脂酸酰胺(ebs)等中的至少一种。所述脂肪酸金属皂类润滑剂例如可以为硬脂酸铅、硬脂酸钙、硬脂酸镁、合成醋酸钙等中的至少一种。所述烷烃及氧化烷烃类润滑剂例如可以为液体石蜡、固体石蜡、聚乙烯蜡、聚丙烯蜡、氧化乙烯蜡等中的至少一种。所述微纳米粒子类润滑剂例如可以为粉末橡胶和/或硅胶微粒。此外,以所述组分a、组分b和组分c的总重量为100重量份计,所述润滑剂的含量可以为0.05-5重量份,优选为0.5-3重量份。
当将该聚乙烯基础树脂制成聚乙烯发泡珠粒时,通常需要加入发泡剂例如二氧化碳。为了提高发泡剂的浸入率和扩散速率、增加泡孔均匀度,优选地,所述聚乙烯基础树脂中还含有泡孔控制剂。所述泡孔控制剂的实例包括但不限于:丙三醇、聚乙二醇、c12-c23的脂肪酸的甘油酯等亲水性化合物。其中,所述聚乙二醇是指具有乙二醇聚合而得的结构的非离子性水溶性聚合物,其数均分子量可以为5万以下,优选为500-6000,更优选为800-4000。另外,所述c12-c23的脂肪酸的甘油酯优选为由硬脂酸和丙三醇形成的单酯、二酯、和三酯中的至少一种。泡孔控制剂的使用能够容易得到高发泡倍率的聚乙烯发泡珠粒。从以低添加量就可得到高发泡倍率的聚乙烯发泡珠粒以及形成模内发泡珠粒成型体时的表观层熔合程度良好、外观优异方面考虑,所述泡孔控制剂优选为丙三醇和/或聚乙二醇,最优选为丙三醇。此外,以所述组分a、组分b和组分c的总重量为100重量份计,所述泡孔控制剂的用量优选为0.1-2重量份,更优选为0.2-0.5重量份。
此外,所述聚乙烯基础树脂中还可以含有现有的各种能够在聚乙烯发泡珠粒中通常使用的其他助剂,例如,抗氧剂、紫外吸收剂、防静电剂、阻燃剂、金属失活剂、颜料、核化剂、填料、稳定剂、增强剂等。上述助剂的种类和含量均可以为本领域的常规选择,对此本领域技术人员均能知悉,在此不作赘述。
所述聚乙烯基础树脂可以按照现有的各种方法制备得到,例如,可以先分别制备组分a、组分b和组分c,然后将所述组分a、组分b和组分c以及任选的其他助剂按照配比在机械混合设备中进行机械混合,接着加入熔融共混设备中进行熔融共混。其中,所述机械混合设备例如可以为高速搅拌机、捏合机等。所述熔融共混设备例如可以为双螺杆挤出机、单螺杆挤出机、开炼机、密炼机等。
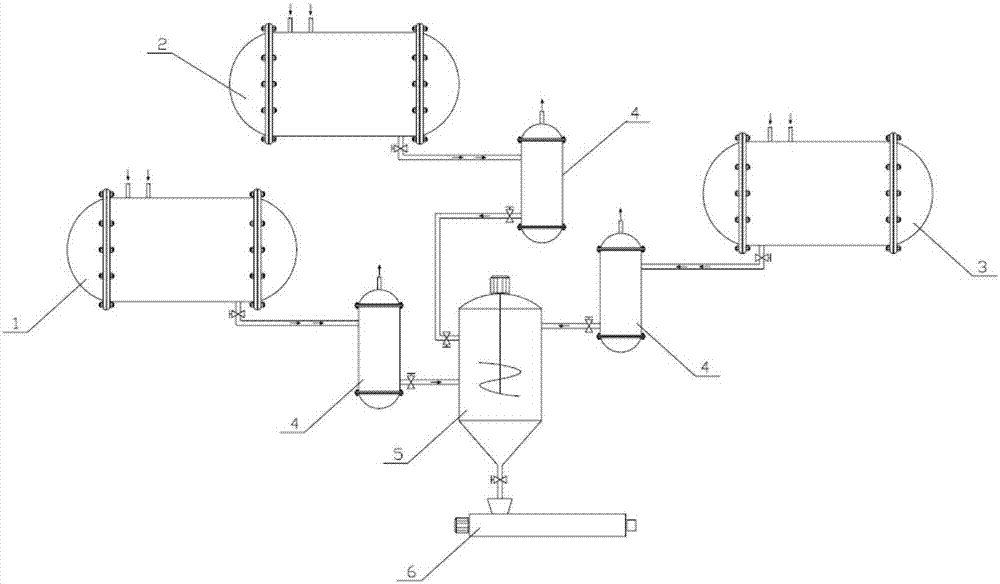
在本发明的一些优选的实施方式中,所述聚乙烯基础树脂在如图1所示的多反应器并联装置中制备得到,所述多反应器并联装置包括第一反应器1、第二反应器2、第三反应器3、固/液(气)分离器4、均化料仓5和熔融造粒系统6,其中,所述第一反应器1、第二反应器2和第三反应器3并联连接,所述固/液(气)分离器4的个数为三个,分别与第一反应器1、第二反应器2和第三反应器3连通,由第一反应器1制备的组分a、由第二反应器2制备的组分b以及由第三反应器3制备的组分c分别在不同的固/液(气)分离器4中进行相分离,然后将经相分离后的组分a、组分b和组分c按比例输送至均化料仓5中并与任选的其他添加剂一起混合均匀,之后送入熔融造粒系统6中进行挤出造粒。其中,各反应器中的聚合可以是间歇聚合,也可以是连续聚合。当采用多反应器并联聚合时,下文中的wa、wb和wc为各组分在相应反应器中的单位时间产量。
根据本发明所述的阻燃聚乙烯组合物,所述氧化膦具有如下式i所示的结构:
式i中,r1、r2和r3相同或不同,各自独立地选自c1-c18直链烷基,c3-c18支链烷基,c1-c18直链烷氧基,c3-c18支链烷氧基,c6-c20取代或未取代芳香基,以及c6-c20取代或未取代的芳氧基。
根据本发明的优选的实施方式,r1、r2和r3各自独立地选自甲基、乙基、丙基、c4-c18直链或支链烷基和c6-c20取代或未取代芳香基;更优选选自c4-c18直链或支链烷基和c6-c18取代或未取代芳香基。
根据本发明的更优选的实施方式,r1、r2和r3各自独立地选自c4-c18直链或支链烷基和碳环数为1或2个的c6-c18芳香基。
根据本发明的进一步优选的实施方式,r1、r2和r3各自独立地选自主碳链具有6个以上的碳原子的c6-c12直链或支链烷基和取代或未取代苯基。
根据本发明,所述芳香基可以带有羟基、羧基等取代基。
根据本发明的进一步优选的实施方式,r1、r2和r3为相同取代基。具有该结构的氧化膦与过渡金属具有更强的配合能力。
根据本发明的优选实施方式,所述氧化膦选自三苯基氧化膦、双(4-羟基苯基)苯基氧化膦、双(4-羧基苯基)苯基氧化膦、三丁基氧化膦、三己基氧化膦、三辛基氧化磷和三癸基氧化膦中的至少一种,更优选选自三苯基氧化膦、三辛基氧化膦、三己基氧化膦和三癸基氧化膦中的至少一种。
根据本发明,所述过渡金属盐包括过渡金属有机盐和/或过渡金属无机盐,优选过渡金属的硝酸盐、硫氰酸盐、甲酸盐、乙酸盐和草酸盐中的至少一种,更优选硝酸盐。所述过渡金属优选为viii族金属元素,更优选为钴和/或镍。具体地,所述过渡金属盐例如选自乙酸钴、乙酸镍、硝酸钴、硝酸镍、硫氰酸镍、硫氰酸钴、硝酸镍和硝酸钴中的至少一种。
根据本发明的一个优选实施方式,所述过渡金属盐为硝酸钴和/或硝酸镍。这两种盐更易于与氧化膦形成配合物,得到更高的产率。
根据本发明的优选实施方式,所述氧化膦与过渡金属盐形成的配合物具有如下结构式ii:
式ii中,m为过渡金属。r1、r2与r3与式i中的r1、r2与r3相同。
r4和r5相同或不相同,各自独立地选自甲酸根离子(hcoo-)、乙酸根离子(ch3coo-)、草酸根离子(c2o4h-)、硝酸根离子(no3-)和硫氰酸根离子(scn-)中的至少一种;优选硝酸根离子和硫氰酸根离子;更优选硝酸根离子。
根据本发明所述的阻燃聚乙烯组合物,其中配合物的制备步骤包括:将1-10重量份、优选2-5重量份的氧化膦与3-15重量份、优选5-10重量份的过渡金属盐在有机溶剂中搅拌混合,然后微波加热,超临界干燥,得到所述配合物;所述有机溶剂优选为乙醇、丙酮、吡啶、四氢呋喃和dmf中的至少一种。
其中,搅拌的速度可以例如为90-120rpm,微波的功率为35-55w,微波加热的温度为35-50℃,加热时间为3-4.5个小时。
在本发明的优选实施方式中,超临界干燥后得到的配合物可表示为m(cho2)2(opr3)2,其中,m可以是ni或者co,r可以是苯基、己基、辛基或癸基。
根据本发明所述的阻燃聚乙烯组合物,基于100重量份的聚乙烯基础树脂,所述阻燃剂的量为5-50重量份,优选10-20重量份;任选地,所述抗氧剂的量为0.1-0.5重量份,优选0.15-0.25重量份。
根据本发明所述的阻燃聚乙烯组合物,所述阻燃剂中进一步包括无机阻燃剂组分,优选所述无机阻燃剂组分选自iia和iiia族金属氢氧化物,更优选选自氢氧化镁和/或氢氧化铝。通过增加无机阻燃剂组分,可进一步增强阻燃效果。
根据本发明的优选实施方式,所述阻燃剂中的配合物与所述无机阻燃剂组分的重量比为(1-5):1,优选为(2.5-3.5):1。
在本发明的一些优选的实施方式中,所述阻燃剂包括:1-10重量份、优选2-5重量份的氧化膦与3-15重量份、优选5-10重量份的过渡金属盐形成的配合物,以及1-10重量份、优选3-6重量份的无机阻燃剂组分。
当包含无机阻燃剂组分时,本发明所述阻燃剂可以通过先制备所述配合物,然后将所述配合物与无机阻燃剂组分进行物理混合来制备得到。这里的物理混合可以是球磨、机械搅拌。优选机械搅拌匀化,搅拌速度为10rpm左右。
本发明所述的阻燃聚乙烯组合物尤其适用于热塑性发泡材料或其成型体的制备中,能够与抗静电剂形成协同促进所用,使热塑性制品达到环保安全的要求,提高阻燃效率。
根据本发明所述的阻燃聚乙烯组合物,所述阻燃聚乙烯组合物中进一步包含碳纳米纤维抗静电剂(导电填料)。优选地,基于100重量份的聚乙烯基础树脂,所述碳纳米纤维抗静电剂的量为0.1-10重量份,优选1-3重量份。
在本发明的一些优选的实施方式中,所述阻燃剂与所述碳纳米纤维抗静电剂的重量比为(3-20):1,更优选(6-15):1。
根据本发明所述的阻燃聚乙烯组合物,优选地,所述阻燃聚乙烯组合物中含有过渡金属(如镍或钴)1-5wt%,例如2-4wt%。这部分过渡金属可以来自所述碳纳米纤维抗静电剂制备过程中所使用的催化剂。作为本发明的一个优势在于,所述使用的碳纳米纤维不需去除其中的过渡金属催化剂,而直接用于制备所述阻燃热塑性材料。由于过渡金属的存在以及其他潜在的原因,本发明使用的碳纳米纤维可以和阻燃剂发生协同效应,有助于生成阻隔火焰和材料的致密碳层,从而可以减少阻燃剂的添加量,且与阻燃剂复合后不互相负面影响导致彼此性能减低,不影响后续的发泡过程及泡沫结构与物理性能。
根据本发明,所述碳纳米纤维的纯度、长径比、直径及形貌并没有特殊要求。
适用于本发明的碳纳米纤维的制备方法包括:将碳源进行酸处理,然后与过渡金属催化剂形成复合物,将所述复合物进行炭化处理。
以下为碳纳米纤维的示例性的制备方法。
1)将碳源用磷酸硝酸盐酸(体积比1:1:1)混合酸处理方法或研磨处理方法进行预处理,得到预处理物。
其中,碳源为凝聚态碳源,可以是碳沥青、石油沥青、煤沥青、煤焦油、天然石墨、人造石墨、竹炭、炭黑、活性炭和石墨烯的至少一种;这里优选碳含量为80wt%以上的碳源,例如碳含量为80wt%以上的煤沥青、石油沥青和竹炭中的至少一种。
2)复合:将预处理物与金属催化剂复合得到复合物。
金属催化剂优选为过渡金属的硫酸盐、硝酸盐、醋酸盐或茂基化合物,所述过渡金属优选为viii族金属元素,例如fe、co或ni,也可以是cr。
在金属催化剂中过渡金属原子与碳源的质量百分比为(35-70):100。
考虑到催化剂中含有氮元素会有利于协同作用促进阻燃效果,这里金属催化剂优选硝酸钴和/或硝酸镍。
3)炭化处理:将复合物在800-1200℃的温度、高纯氮气保护下进行炭化反应,恒温0.5-5小时,降温到室温得到自组装碳纤维。这里炭化处理的温度优选950-1150℃,恒温反应1.5-2.5小时。无需后处理去除金属杂质。
与现有技术中常用的短效抗静电剂,如高分子聚合物抗静电剂相比,本发明使用的碳纳米纤维是一种长效抗静电剂,能够提供长效的抗静电作用。
所述阻燃聚乙烯组合物可以按照现有的各种方法制备得到,例如,直接将聚乙烯基础树脂和所述阻燃剂、抗静电剂以及选择性含有的润滑剂和其他助剂按照配比在机械混合设备中进行机械混合,然后加入熔融共混设备中在170-200℃条件下进行熔融共混造粒。也可以先将少量聚乙烯分别和阻燃剂或导电填料浓缩共混,在170-210℃条件下制得阻燃母粒和抗静电母粒,再将两种母粒与聚乙烯按比例掺混,在170-200℃条件下进行造粒。其中,所述机械混合设备例如可以为高速搅拌机、捏合机等。所述熔融共混设备例如可以为双螺杆挤出机、单螺杆挤出机、开炼机、密炼机、buss捏合机等。
本发明第二方面提供了一种阻燃聚乙烯发泡珠粒,其通过将包含100重量份的如本发明第一方面所述的阻燃聚乙烯组合物,0.001-1重量份、优选0.01-0.05重量份的泡孔成核剂以及0.2重量份的抗氧剂的物料,经过浸渍发泡工艺制备得到。
在本发明的一些实施方式中,所述泡孔成核剂的种类可以为本领域的常规选择,例如,可以选自硼酸锌、二氧化硅、滑石粉、碳酸钙、硼砂和氢氧化铝中的至少一种。从原料易得的角度出发,所述泡孔成核剂特别优选为滑石粉。
此外,本发明还提供了上述阻燃聚乙烯发泡珠粒的制备方法,该方法包括将上述阻燃聚乙烯组合物进行造粒,并将得到的聚乙烯颗粒进行发泡。
所述造粒可以采用现有的各种方式进行,例如,可以将阻燃聚乙烯组合物经由双螺杆或单螺杆挤出机的一个或多个模头挤塑成线材并切割而获得阻燃聚乙烯珠粒,也可以使用水下微颗粒切粒系统,具体操作过程为本领域技术人员公知。
根据本发明的一些具体实施方式,所述造粒按照如下方式进行:将上述阻燃聚乙烯组合物利用高速搅拌机共混后,通过双螺杆挤出机挤出,热切后导入75℃以下、优选70℃以下、更优选55-65℃的水中进行微颗粒切割,使每个颗粒的长度/直径比为0.5-2.0、优选为0.8-1.3、更优选为0.9-1.1,且平均重量为0.1-20mg、优选为0.2-10mg、更优选为1-3mg。此处所述的长度/直径比为200个任意选择的聚乙烯颗粒的平均值。
所述发泡也可以采用现有的各种方式进行,例如,可以采用挤出发泡法进行,也可以采用反应釜浸渍发泡法进行,优选采用反应釜浸渍发泡法进行这样得到的聚乙烯发泡珠粒为非交联结构,从而可以按照聚乙烯改性材料回收利用,不造成二次污染,符合循环经济的要求。
根据本发明的一些具体实施方式,所述发泡采用反应釜浸渍发泡法进行,具体过程如下:
(1)在高压釜中,将聚乙烯颗粒与分散介质、表面活性剂、分散剂和分散增强剂等助剂混合均匀;
(2)先盖紧釜盖,用排空气法即使用发泡剂将高压釜内残余空气排出,之后将发泡剂继续喂入该高压釜中,开始加热并初步调整压力直到其稳定,随后搅拌该高压釜,搅拌速度为50-150rmp、优选为90-110rmp,以匀速将其加热到比膨胀稳定低0.1-5℃、优选低0.5-1℃的温度;
(3)调整高压釜内压力达到发泡所需压力,该压力为1-10mpa、优选为3-5mpa,以0.1℃/分钟的平均加热速度将温度升高到发泡温度,发泡温度比微颗粒熔融温度低0.1-5℃、优选低0.5-1℃,在发泡温度和压力条件下,持续搅拌0.1-2小时、优选0.25-0.5小时;
(4)将高压釜的出料口打开,使高压釜内的物料排泄到收集罐中,以获得聚乙烯发泡珠粒,在进行出料的同时喂入二氧化碳气体,使得在全部粒子完全发泡且进入收集罐前,将高压釜中的压力保持在发泡压力附近。
在本发明中,所述压力均指表压。
所述分散介质可以为现有的各种能够使聚乙烯颗粒分散于其中而不溶解该聚乙烯颗粒的组分,例如,可以为水、乙二醇、甘油、甲醇、乙醇等中的至少一种,特别优选为水。此外,相对于100重量份的聚乙烯颗粒,所述分散介质的用量可以为1000-5000重量份,优选为2500-3500重量份。
所述表面活性剂可以为现有的各种能够促进聚乙烯颗粒分散在分散介质中的组分,例如,可以为硬脂酸、十二烷基苯磺酸钠、季铵化物、卵磷脂、氨基酸、甜菜碱、脂肪酸甘油酯、脂肪酸山梨坦、聚山梨酯等中的至少一种,特别优选为十二烷基苯磺酸钠。此外,相对于100重量份的聚乙烯颗粒,所述表面活性剂的用量可以为0.001-10重量份,优选为0.01-5重量份,更优选为0.1-0.5重量份。
所述分散剂添加的目的是为了防止聚乙烯颗粒在发泡期间彼此熔融粘合。所述分散剂可以为有机分散剂,也可以为无机分散剂,优选为无机分散剂。所述无机分散剂可以是天然的或合成的粘土矿物(例如高岭土、云母、镁铝榴石、粘土等)、矾土、二氧化钛、碱式碳酸镁、碱式碳酸锌、碳酸钙、二氧化硅、硼酸锌和氧化铁等中的至少一种,特别优选为高岭土。此外,相对于100重量份的聚乙烯颗粒,所述分散剂的用量可以为0.01-20重量份,优选为0.1-10重量份,更优选为0.5-5重量份。
所述分散增强剂添加的目的是为了提高分散剂的分散效率,即在减少分散剂用量的同时保留其防止颗粒间熔融粘合的功能。所述分散增强剂可以为现有的各种在100ml、40℃水中溶解度为1mg并提供二价或三价阴离子或者阳离子的无机化合物。所述分散增强剂的实例包括但不限于氮化镁、硝酸镁、磷酸铝、硫酸镁、氮化铝、硝酸铝、硫酸铝、氯化铁、硫酸铁和硝酸铁等中的至少一种,优选为硫酸铝。所述分散增强剂的使用有利于得到表观密度为100g/l以上的聚乙烯发泡珠粒。此外,相对于100重量份的聚乙烯颗粒,所述分散增强剂的用量可以为0.0001-1重量份,优选为0.01-0.2重量份。
所述发泡剂可以为有机类物理发泡剂,也可以为无机类物理发泡剂。其中,所述有机类物理发泡剂的实例包括但不限于脂肪族烃类例如丙烷、丁烷、戊烷、己烷和庚烷,脂环族烃类例如环丁烷和环己烷,以及卤代烃类例如氯氟甲烷、三氟甲烷、1,2-二氟乙烷、1,2,2,2-四氟乙烷、甲基氯、乙基氯和二氯甲烷等中至少一种。所述无机类物理发泡剂的实例包括但不限于空气、氮气、二氧化碳、氧气和水中的至少一种。考虑到聚乙烯发泡珠粒表观密度的稳定性(均一性)、低成本和环境友好问题,所述发泡剂优选为二氧化碳和/或氮气,特别优选为二氧化碳。此外,所述发泡剂的用量可以根据发泡剂的具体种类、发泡温度以及所要生产的聚乙烯发泡珠粒的表观密度来进行确定。例如,当采用氮气作为发泡剂且采用水作为分散介质时,发泡装置泄压时该密闭容器内的压力(即该密闭容器内上部空间中的压力(表压))控制在1-12mpa;当使用二氧化碳作为发泡剂时,则将上述表压控制在1-7mpa。一般来说,该密闭容器内上部空间中的理想压力随要得到的聚乙烯发泡珠粒的表观密度降低而增大。
利用本发明方法制备的聚乙烯发泡珠粒具有成本低、泡孔致密及孔径分布均匀等优点,可应用于汽车部件、食品及电子包装以及建筑装饰等对塑料制品轻量化有较高要求的场合。
本发明第三方面提供了一种阻燃聚乙烯成型体,其由根据本发明第二方面所述的阻燃聚乙烯发泡珠粒经模塑成型工艺制备得到。
根据本发明所述的阻燃聚乙烯成型体,所述模塑成型可以在现有的各种模塑成型机中进行,并且模塑成型的条件均可以为本领域的常规选择,对此本领域技术人员均能知悉,在此不作赘述。
本发明所述用语“基础树脂”是指纯树脂,即未形成组合物的树脂。
本发明所述用语“无卤”是指化合物或混合物或组合物中不含卤素。
本发明所述用语“任选地”是指含有或不含有,亦指加入或不加入。
与现有技术相比,本发明具有以下有益效果:
(1)本发明提供了一种无卤阻燃剂,该阻燃剂中包含无机阻燃剂组分和一种长效抗静电剂,这两种功能化助剂发挥协同作用,可以有效提高阻燃效率,改善阻燃效果,降低阻燃剂添加量,同时对抗静电性能没有负面影响。
(2)本发明提供的阻燃聚乙烯发泡珠粒具有抗冲、较低的成型温度、抗静电、阻燃性能优良等优点,因而适用于汽车部件、医疗器械、电子包装、家居用品、低温冷链包装、体育器材、建筑保温及航空航天等对阻燃抗静电低温抗冲击有综合性要求的领域的优良材料。本发明提供的阻燃聚乙烯组合物的制备方法简单有效、易于操作。
(3)本发明采用二氧化碳作为发泡剂,与现有技术中使用有机类发泡剂相比,具有环境友好、安全等优点。
(4)本发明制备的发泡阻燃聚乙烯珠粒为非交联结构,可以按照一般聚乙烯改性材料回收利用,不造成二次污染,符合循环经济的要求。
附图说明
下面结合附图来对本发明作进一步详细说明。
图1显示了用于制备聚乙烯基础树脂的多反应器并联装置的结构示意图;图中附图标记的含义如下:1-第一反应器;2-第二反应器;3-第三反应器;4-固/液(气)分离器;5-均化料仓;6-熔融造粒系统。
图2显示了本发明实施例1的阻燃聚乙烯发泡珠粒的截面扫描电镜照片。
图3显示了本发明对比例1的传统阻燃聚乙烯发泡珠粒的截面扫描电镜照片。
具体实施方式
参考下列实施例进一步详细描述本发明,但是应该说明:本发明决不局限于这些实施例。
本发明实施例中的有关数据按以下测试方法获得:
(1)分子量分布宽度指数mw/mn:采用英国polymerlaboratories公司生产的型号为pl-gpc220型凝胶渗透色谱仪结合ir5型红外检测器测定,其中凝胶色谱仪的色谱柱为3根串联的plgel10μmmixed-b柱,溶剂及流动相为1,2,4-三氯苯(含0.3g/1000ml抗氧剂2,6-二叔丁基对甲酚),柱温为150℃,流速为1.0ml/min,采用pl公司生产的easicalps-1窄分布聚苯乙烯标样进行普适标定;
(2)熔融指数mi:按照gb/t3682-2000中规定的方法进行测定,其中,测试温度为190℃,载荷为2.16kg;
(3)密度:按照gb/t1033.2-2010中规定的方法并采用密度梯度柱法进行测定;
(4)极限氧指数测试:按照国标gb/t5454-1997描述的方法进行测试;
(5)表面电阻率测试:按照gb/t1410-2006进行测试。
实施例
实施例1
本实施例制备的阻燃剂、阻燃聚乙烯组合物和发泡珠粒等产品的原料配比和反应条件列于表1和表2中,表2还列出了发泡珠粒的性能参数。表中,阻燃剂组分a是氧化膦,阻燃剂组分b是过渡金属盐,阻燃剂组分c是无机阻燃剂组分。
(一)阻燃剂的制备
将7重量份的三苯基氧化膦与3重量份的硝酸钴加入乙醇中,以100rpm的速率进行搅拌,然后在搅拌下使用微波加热混合物料,加热功率为50w,温度为40℃,反应时间4h。将微波加热反应后的物料进行超临界干燥,得到三苯基氧化膦与硝酸钴的形成的配合物co(opph3)2(no3)2。
将上述制备的配合物co(opph3)2(no3)2与5重量份的氢氧化镁机械搅拌匀化,搅拌速度为10rpm,得到阻燃剂。
(二)碳纳米纤维抗静电剂的制备
以碳含量为80mol%以上的煤沥青为碳源,用磷酸/硝酸/盐酸(体积比1:1:1)混合酸进行研磨预处理,得到预处理物。
将上述预处理物和催化剂硝酸钴加入球磨机中混合,得到复合物。
将所述复合物在950℃高纯氮气保护下进行炭化反应,恒温1.5小时,然后降温到室温,得到自组装碳纳米纤维。无需后处理去除催化剂金属杂质,经测定,含钴2.0wt%。
(三)聚乙烯基础树脂pe301的制备
本实施例提供的聚乙烯基础树脂含有组分a、组分b、组分c、润滑剂和泡孔控制剂。其中,组分a、组分b和组分c均为乙烯/α烯烃共聚的线性低密度聚乙烯(lldpe),其中,组分a和组分b采用茂金属催化剂制备得到(所述茂金属催化剂体系为由cn102453124a实施例1制备得到的负载型茂金属催化剂,下同),组分c采用齐格勒-纳塔催化剂制备得到(所述齐格勒-纳塔催化剂体系为由cn101838351a实施例1制备得到的齐格勒-纳塔催化剂体系,下同)。具体步骤如下:
将乙烯、α烯烃、氢气和氮气(乙烯、α烯烃、氢气和氮气均为聚合级,经脱除水、氧后使用,下同)加入到流化床气相反应器中,然后加入催化剂体系,之后在温度为84-88℃、压力为1.8-2.0mpa的条件下聚合,分别得到组分a、组分b和组分c。其中,组分a、组分b和组分c的熔融指数的控制通过调节氢气的加入量而实现,密度的控制通过调节α烯烃的种类和加入量而实现。制备组分a的过程中所用α烯烃为1-己烯,制备组分b的过程中所用α烯烃为1-己烯,制备组分c的过程中所用α烯烃为1-丁烯。
经检测,由上述方法制备得到的组分a、组分b和组分c的性能如下:
组分a的熔融指数mia=1.5g/10min,密度ρa=0.913g/cm3,分子量分布指数mw/mn=3.4,α烯烃共聚单体的摩尔含量=7.5mol%;
组分b的熔融指数mib=2.1g/10min,密度ρb=0.913g/cm3,分子量分布指数mw/mn=3.2,α烯烃共聚单体的摩尔含量=7.5mol%;
组分c的熔融指数mic=15g/10min,密度ρc=0.905g/cm3,分子量分布指数mw/mn=4.6,α烯烃共聚单体的摩尔含量=10.1mol%。
润滑剂为由瑞士科莱恩公司生产的peg润滑剂,数均分子量为10000。
泡孔控制剂为北京化学试剂公司生产的丙三醇。
将上述组分a、组分b和组分c按配比进行称重并混合,其中组分a的质量份数wa为80重量份,组分b的质量份数wb为10重量份,组分c的质量份数wc为20重量份,wa/wc=4(满足5.2×lgmia+11.6≥wa/wc≥0.9×lgmia+2.1,也满足2.9×lgmia+6.8≥wa/wc≥1.1×lgmia+2.7);然后加入润滑剂和泡孔控制剂(以上述组分a、组分b和组分c的总重量为100重量份计,润滑剂的加入量为0.1重量份,泡孔控制剂的加入量为0.4重量份),之后将混合物加入到高速搅拌器中混合均匀,再将混合好的物料加入到南京科倍隆公司制造的双螺杆挤出机的喂料器中,物料经由喂料器进入双螺杆中,加工过程中螺杆的温度保持在160-210℃之间,经螺杆熔融混合均匀后挤出,切粒并烘干,得到聚乙烯基础树脂粒料,经检测其熔融指数mi=2.4g/10min。
(四)阻燃聚乙烯组合物的制备
将上述各组分按配比进行称重并混合,其中步骤(三)中的聚乙烯基础树脂为100重量份,阻燃剂组分为15重量份,碳纳米纤维抗静电剂为1重量份,泡孔成核剂滑石粉(大连富士矿产公司生产,粒径分布为20-30μm)为0.25重量份。此外,该组合物的制备过程中还加入了加工助剂包括抗氧剂1010(basf公司)、抗氧剂168(basf公司)等,用量为常规用量,相对于聚乙烯基础树脂的100重量份,分别为0.2及0.1重量份。之后将混合物加入到高速搅拌器中混合均匀,再将混合好的物料加入到科倍隆公司制造的双螺杆挤出机的喂料器中,物料经由喂料器进入双螺杆中,加工过程中螺杆的温度保持在170-200℃之间,经螺杆熔融混合均匀、进入lab100微粒子制备系统,扭矩控制在65%左右,转速300rpm。得到阻燃聚乙烯组合物微颗粒。
(五)阻燃聚乙烯发泡珠粒的制备方法
将步骤(四)得到的阻燃聚乙烯组合物微颗粒与分散介质去离子水、表面活性剂十二烷基苯磺酸钠、分散剂高岭土和分散增强剂硫酸铝等助剂一次性加入高压釜中混合均匀,且相对于100重量份的阻燃聚乙烯组合物,分散介质的用量为2700重量份,表面活性剂的用量为0.4重量份,分散剂的用量为5重量份,分散增强剂的用量为0.2重量份。
盖紧高压釜釜盖,使用二氧化碳将高压釜内残余空气排出,之后将二氧化碳继续喂入高压釜中,开始加热并初步调整釜内压力直到其稳定,随后搅拌该高压釜,搅拌速度为100rmp,以匀速将高压釜内的温度加热至119℃。
调整高压釜内压力至4mpa,并以0.1℃/分钟的平均加热速度将温度升高至119.5℃,接着在上述压力和温度下持续搅拌0.5小时。
将高压釜的出料口打开,使高压釜内的物料排泄到收集罐中,以获得发泡珠粒,在进行出料的同时喂入二氧化碳气体,使得在全部粒子完全发泡且进入收集罐前,将高压釜中的压力保持在发泡压力附近。
收集珠粒后脱水干燥,使用孔径为3.35mm和2.8mm的筛子筛分出粒径为2.8-3.35mm的聚乙烯发泡珠粒,其表面的电镜照片和截面的电镜照片分别如图2和图3所示。从图2和图3的结果可以看出,由本发明提供的聚乙烯组合物获得的聚乙烯发泡珠粒的泡孔致密均匀,表面光滑,泡孔尺寸较小。
(六)阻燃聚乙烯发泡珠粒成型体的制备
将步骤(五)得到的阻燃聚乙烯发泡珠粒使用模塑成型机(德国kurtzersa公司生产的kurtzt-line,下同)在0.15mpa的压力下模塑成型,随后将所获成型体在温度为65℃、压力为标准大气压的条件下熟化24小时,即得到模塑成型品。具体的发泡珠粒熔合压力等模塑参数见表2。模塑成型品用于氧指数、残炭率、火焰高度、烟雾情况、表面电阻率、压缩强度的测试。按照国标gbt2406.2-2009描述的方法进行氧指数测试,按照gb/t1410-2006进行表面电阻率测试。从发泡珠粒成型体中切割出50*50*25mm的试样,基于美国astm标准d3575-08进行压缩强度测试,利用10mm/min的压缩速度进行压缩试验,得到成型体被压缩50%时的压缩强度。上述各项测试的结果见表2。
实施例2
(一)阻燃剂的制备方法和(二)碳纳米纤维抗静电剂的制备方法同实施例1,不同之处在于,表1和表2中所示的原料配方和反应条件。例如,该实施例形成的阻燃剂为三辛基氧化膦与硝酸镍形成的配合物ni(opot3)2(no3)2。
(三)聚乙烯基础树脂pe302的制备
本实施例提供的聚乙烯基础树脂含有组分a、组分b、组分c、润滑剂和泡孔控制剂。其中,组分a、组分b和组分c均为乙烯/α烯烃共聚的线性低密度聚乙烯(lldpe),其中,组分a和组分b采用茂金属催化剂制备得到,组分c采用齐格勒-纳塔催化剂制备得到。具体步骤如下:
将乙烯、α烯烃、氢气和氮气加入到流化床气相反应器中,然后加入催化剂体系,之后在温度为84-88℃、压力为1.8-2.0mpa的条件下聚合,分别得到组分a、组分b和组分c。其中,组分a、组分b和组分c的熔融指数的控制通过调节氢气的加入量而实现,密度的控制通过调节α烯烃的种类和加入量而实现。制备组分a的过程中所用α烯烃为1-丁烯,制备组分b的过程中所用α烯烃为1-丁烯,制备组分c的过程中所用α烯烃为1-己烯。
经检测,由上述方法制备得到的组分a、组分b和组分c的性能如下:
组分a的熔融指数mia=0.01g/10min,密度ρa=0.930g/cm3,分子量分布指数mw/mn=3.0,α烯烃共聚单体的摩尔含量=1.6mol%;
组分b的熔融指数mib=9.0g/10min,密度ρb=0.930g/cm3,分子量分布指数mw/mn=2.8,α烯烃共聚单体的摩尔含量=1.9mol%;
组分c的熔融指数mic=40g/10min,密度ρc=0.922g/cm3,分子量分布指数mw/mn=4.4,α烯烃共聚单体的摩尔含量=4.0mol%。
润滑剂为由瑞士科莱恩公司生产的peg润滑剂,数均分子量为10000。
泡孔控制剂为北京化学试剂公司生产的丙三醇。
将上述组分a、组分b和组分c按配比进行称重并混合,其中组分a的质量份数wa为55重量份,组分b的质量份数wb为5重量份,组分c的质量份数wc为55重量份,wa/wc=1(满足5.2×lgmia+11.6≥wa/wc≥0.9×lgmia+2.1,也满足2.9×lgmia+6.8≥wa/wc≥1.1×lgmia+2.7);然后加入润滑剂和泡孔控制剂(以上述组分a、组分b和组分c的总重量为100重量份计,润滑剂的加入量为0.1重量份,泡孔控制剂的加入量为0.5重量份),之后将混合物加入到高速搅拌器中混合均匀,再将混合好的物料加入到南京科倍隆公司制造的双螺杆挤出机的喂料器中,物料经由喂料器进入双螺杆中,加工过程中螺杆的温度保持在180-240℃之间,经螺杆熔融混合均匀后挤出,切粒并烘干,得到聚乙烯基础树脂粒料,经检测其熔融指数mi=0.7g/10min。
(四)阻燃聚乙烯组合物的制备
阻燃聚乙烯组合物的制备同实施例1,不同之处在于表1中的原料配比。例如,该实施例中的阻燃剂组分为14重量份,碳纳米纤维抗静电剂为1.5重量份,泡孔成核剂滑石粉为0.35重量份。
(五)阻燃聚乙烯发泡珠粒的制备
将步骤(四)得到的阻燃聚乙烯组合物微颗粒与分散介质去离子水、表面活性剂十二烷基苯磺酸钠、分散剂高岭土和分散增强剂硫酸铝等助剂一次性加入高压釜中混合均匀,且相对于100重量份的聚乙烯组合物粒料,分散介质的用量为3000重量份,表面活性剂的用量为0.3重量份,分散剂的用量为4.5重量份,分散增强剂的用量为0.15重量份。
盖紧高压釜釜盖,使用二氧化碳将高压釜内残余空气排出,之后将二氧化碳继续喂入高压釜中,开始加热并初步调整釜内压力直到其稳定,随后搅拌该高压釜,搅拌速度为100rmp,以匀速将高压釜内的温度加热至128.5℃。
调整高压釜内压力至4mpa,并以0.1℃/分钟的平均加热速度将温度升高至129℃,接着在上述压力和温度下持续搅拌0.5小时。
将高压釜的出料口打开,使高压釜内的物料排泄到收集罐中,以获得发泡珠粒,在进行出料的同时喂入二氧化碳气体,使得在全部粒子完全发泡且进入收集罐前,将高压釜中的压力保持在发泡压力附近。
收集珠粒后脱水干燥,使用孔径为3.35mm和2.8mm的筛子筛分出粒径为2.8-3.35mm的聚乙烯发泡珠粒。
(六)聚乙烯发泡珠粒成型体的制备
将步骤(五)得到的聚乙烯发泡珠粒使用模塑成型机在0.19mpa的压力下模塑成型,随后将所获成型体在温度为65℃、压力为标准大气压的条件下熟化24小时,即得到模塑成型品。对其进行氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试,其结果见表2。
实施例3
(一)阻燃剂的制备方法和(二)碳纳米纤维抗静电剂的制备方法同实施例1,不同之处在于,表1和表2中所示的原料配方和反应条件。例如,该实施例形成的阻燃剂为三辛基氧化膦与硝酸钴形成的配合物co(opot3)2(no3)2。
(三)聚乙烯基础树脂pe303的制备
本实施例提供的聚乙烯基础树脂用图1所示的多反应器并联装置聚合得到,其中第一反应器1聚合制备组分a、第二反应器2聚合制备组分b、第三反应器3聚合制备组分c,这三种组分均为乙烯/α烯烃共聚的线性低密度聚乙烯(lldpe),其中,组分a和组分b采用茂金属催化剂制备得到,组分c采用齐格勒-纳塔催化剂制备得到。具体步骤如下:
将α烯烃、正己烷和氢气加入到聚合反应器中,并将聚合反应器加热到预设的聚合温度,之后将乙烯单体和催化剂体系同时加入到聚合反应器中,其中组分a和组分b在温度为140℃、压力为2.5mpa的条件下聚合得到;组分c在温度为240℃、压力为14.8mpa的条件下聚合得到。其中,组分a、组分b和组分c的熔融指数的控制通过调节氢气的加入量而实现,密度的控制通过调节α烯烃的种类和加入量而实现。制备组分a的过程中所用α烯烃为1-辛烯,制备组分b的过程中所用α烯烃为1-丁烯,制备组分c的过程中所用α烯烃为1-丁烯。
在制备过程中将第一反应器1中组分a的单位时间产量wa、第二反应器2中组分b的单位时间产量wb与第三反应器3中组分c的单位时间产量wc的重量比维持在wa:wb:wc=75:2:35,其中wa/wc=2.1(满足5.2×lgmia+11.6≥wa/wc≥0.9×lgmia+2.1,也满足2.9×lgmia+6.8≥wa/wc≥1.1×lgmia+2.7)。
经检测,由上述方法制备得到的组分a、组分b和组分c的性能如下:
组分a的熔融指数mia=0.1g/10min,密度ρa=0.920g/cm3,分子量分布指数mw/mn=3.1,α烯烃共聚单体的摩尔含量=2.1mol%;
组分b的熔融指数mib=5.0g/10min,密度ρb=0.920g/cm3,分子量分布指数mw/mn=3.5,α烯烃共聚单体的摩尔含量=5.1mol%;
组分c的熔融指数mic=25g/10min,密度ρc=0.920g/cm3,分子量分布指数mw/mn=4.2,α烯烃共聚单体的摩尔含量=5.7mol%。
润滑剂为由瑞士科莱恩公司生产的peg润滑剂,数均分子量为10000。
泡孔控制剂为北京化学试剂公司生产的丙三醇。
将上述组分a、组分b和组分c按单位时间产量比例分别输送到不同的固/液(气)分离器4中进行相分离并进而输送到带有搅拌的均化料仓5中,然后按配比加入润滑剂和泡孔控制剂进行混合均化。其中,以上述组分a、组分b和组分c的总重量为100重量份计,润滑剂的加入量为0.1重量份,泡孔控制剂的加入量为0.5重量份。之后将经均化料仓5均化的混合物加入到南京科倍隆公司制造的双螺杆挤出机的喂料器中,物料经由喂料器进入双螺杆中,加工过程中螺杆的温度保持在160-210℃之间,经螺杆熔融混合均匀后挤出,切粒并烘干,得到聚乙烯基础树脂粒料,经检测其熔融指数mi=0.6g/10min。
(四)阻燃聚乙烯组合物的制备
阻燃聚乙烯组合物的制备同实施例1,不同之处在于表1中的原料配比。例如,该实施例中的阻燃剂组分为15.5重量份,碳纳米纤维抗静电剂为1.5重量份,泡孔成核剂滑石粉为0.45重量份。
(五)阻燃聚乙烯发泡珠粒的制备
将步骤(四)得到的阻燃聚乙烯组合物微颗粒与分散介质去离子水、表面活性剂十二烷基苯磺酸钠、分散剂高岭土和分散增强剂硫酸铝等助剂一次性加入高压釜中混合均匀,且相对于100重量份的聚乙烯组合物粒料,分散介质的用量为3000重量份,表面活性剂的用量为0.29重量份,分散剂的用量为5重量份,分散增强剂的用量为0.14重量份。
盖紧高压釜釜盖,使用二氧化碳该高压釜内残余空气排出,之后将二氧化碳继续喂入高压釜中,开始加热并初步调整釜内压力直到其稳定,随后搅拌该高压釜,搅拌速度为100rmp,以匀速将高压釜内的温度加热至121.5℃。
调整高压釜内压力至5mpa,并以0.1℃/分钟的平均加热速度将温度升高至122℃,接着在上述压力和温度下持续搅拌0.5小时。
将高压釜的出料口打开,使高压釜内的物料排泄到收集罐中,以获得发泡珠粒,在进行出料的同时喂入二氧化碳气体,使得在全部粒子完全发泡且进入收集罐前,将高压釜中的压力保持在发泡压力附近。
收集珠粒后脱水干燥,使用孔径为3.35mm和2.8mm的筛子筛分出粒径为2.8-3.35mm的聚乙烯发泡珠粒。
(六)阻燃聚乙烯发泡珠粒成型体的制备
将步骤(五)得到的聚乙烯发泡珠粒使用模塑成型机在0.18mpa的压力下模塑成型,随后将所获成型体在温度为65℃、压力为标准大气压的条件下熟化24小时,即得到模塑成型品。对其进行氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试,其结果见表2。
实施例4
按照实施例1的方法制备阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体。不同之处在于,步骤(四)中泡孔成核剂滑石粉的用量为0.15重量份,且步骤(六)中的模塑成型压力为0.17mpa,得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
实施例5
按照实施例2的方法制备阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体。不同之处在于,发泡剂为氮气与二氧化碳体积比1:1的混合气,且步骤(六)中的模塑成型压力为0.18mpa,得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
实施例6
按照实施例3的方法制备阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体。不同之处在于,步骤(四)中泡孔成核剂滑石粉的用量为0.1重量份,发泡剂为氮气,且步骤(六)中的模塑成型压力为0.17mpa,得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例1
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,将聚乙烯基础树脂替换为pe302,同时将阻燃剂替换为红磷,且用量为20重量份以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例2
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,将聚乙烯基础树脂替换为pe302,同时将阻燃剂替换为只使用氢氧化镁,且用量为30重量份以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例3
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,将聚乙烯基础树脂替换为pe302,同时将阻燃剂替换为六溴环十二烷与三氧化二锑的组合物,且用量为30重量份以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例4
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,将聚乙烯基础树脂替换为pe302,同时将抗静电剂替换为炭黑,且用量为6重量份以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例5
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,将聚乙烯基础树脂换成普通线性低密度聚乙烯lldpe7042以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例6
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,阻燃剂中不含有阻燃剂组分b硝酸钴以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
对比例7
阻燃剂、碳纳米纤维抗静电剂、聚乙烯基础树脂、阻燃聚乙烯组合物、阻燃聚乙烯发泡珠粒和阻燃聚乙烯发泡珠粒成型体的制备方法同实施例1,不同之处在于,阻燃剂中不含有阻燃剂组分a三苯基氧化膦以及表1和表2中的实验条件。得到的模塑成型品的氧指数、残炭率、火焰高度、烟雾情况、表面电阻率和压缩强度的测试结果见表2。
通过实施例1-6可以看出,本发明制备的pe301、pe302、pe303具有良好的发泡性能,以其作为基础树脂,加入有机磷配合物与无机氢氧化物复配的阻燃剂,含镍或钴的碳纳米纤维作为抗静电剂,制备阻燃抗静电聚乙烯基础树脂。随后,按照本发明提供的釜式浸渍发泡法,通过调整发泡压力及温度等条件可以得到密度为0.08-0.20g/l的发泡珠粒,使用非超临界二氧化碳作为发泡剂时发泡效果良好,泡孔密度较高,泡孔致密均匀,泡孔尺寸较小,泡孔壁较薄,珠粒表面光滑,其中实施例1的珠粒的表面及截面图如附图2所示。具体的模塑成型体的力学、阻燃及抗静电性能如表2所示。其中,良好的泡孔结构使得成型体的压缩性能优良;成型体氧指数及相关阻燃测试情况表明,阻燃剂与抗静电剂可以发挥协同效应,可以有效降低阻燃剂添加量,氧指数高于28,可以用于对阻燃水平要求较高领域,同时表面电阻率达到109ω抗静电级别。
通过对比例1-4及表2可以看出,使用传统的红磷、溴系阻燃剂、单独使用氢氧化镁等阻燃剂与含镍或钴的碳纳米纤维等配合,作为复配的阻燃抗静电剂得到的聚乙烯基础树脂制备发泡珠粒,所得珠粒所形成的成型体的阻燃能力和抗静电性能逊于实施例1-6所述组合物所得到的发泡珠粒,且对比例中所述阻燃剂和抗静电剂的加入对于发泡性能起到负面作用,泡孔不均匀,泡孔壁有破损。将实施例1-6与对比例6-7相比,可以看出,使用有机磷与镍钴等过渡金属形成的配合物及氢氧化镁或氢氧化铝作为阻燃剂与碳纳米纤维组成的抗静电体系中,过渡金属与氢氧化镁发生协同催化作用,提高磷系阻燃剂的阻燃效率。碳纳米纤维可以在树脂内部构建有效的导电网络,从而形成长效的抗静电网络体系,有效的降低了发泡珠粒成型体的表面电阻率。碳纤维中残存的镍或钴系催化剂同样与配合物形成良好的协同作用,提高阻燃效率。此外,在对比例1中,使用传统红磷阻燃剂与抗静电剂形成的体系所得到的组合物,两者没有协同效应,反而互相影响降低了阻燃和抗静电性能,且对珠粒的泡孔结构造成负面影响,所得到的发泡珠粒的泡孔密度较低,泡孔直径较大,且出现泡孔壁破裂的现象(如附图3所示)。
应当注意的是,以上所述的实施例仅用于解释本发明,并不构成对本发明的任何限制。通过参照典型实施例对本发明进行了描述,但应当理解为其中所用的词语为描述性和解释性词汇,而不是限定性词汇。可以按规定在本发明权利要求的范围内对本发明作出修改,以及在不背离本发明的范围和精神内对本发明进行修订。尽管其中描述的本发明涉及特定的方法、材料和实施例,但是并不意味着本发明限于其中公开的特定例,相反,本发明可扩展至其他所有具有相同功能的方法和应用。
- 还没有人留言评论。精彩留言会获得点赞!