基于梅花鹿全基因组开发的STR分子标记及其应用的制作方法
本发明涉及动物资源学、分子生物学和生物信息学领域,具体地说,涉及基于梅花鹿全基因组开发的str分子标记及其应用。
背景技术:
随着各类哺乳动物基因组计划的完成,测序技术和生物信息学分析技术的飞速发展,人们的目光越来越多地集中在特种经济动物遗传资源的评价、保护和利用上,特别是梅花鹿遗传资源。由于野生梅花鹿具有特殊的药用价值,我国在公元前14—12世纪就开始对野生梅花鹿进行家养化利用,是世界上养殖梅花鹿最早的国家,同时也是最早将梅花鹿产品应用于医药保健的国家,主要用于中药和保健品原料。目前我国的野生梅花鹿己十分罕见,仅存在于一些片段化生境中,其分布区正在不断缩减,野生梅花鹿已处于濒危状态,早已被列入中国国家一级保护动物,中国濒危动物红皮书:濒危。对于梅花鹿遗传资源的评价和利用,指的是家养梅花鹿,由野生东北梅花鹿驯化而来。野生梅花鹿经过多年人工驯养,变得温顺,适应性强,生产力和遗传力稳定,成为发展养鹿业的物质基础和有力保证。但由于单纯追求高效益,没有相应的原种保护措施,已使家养梅花鹿6个选育品种和1个品系种质资源的保留受到品种间杂交和种间杂交的影响,这些品种都不同程度退化和改变,有些品种呈现下降或灭绝趋势。此外,梅花鹿养殖过程中,错误的系谱记录非常普遍,对后续的遗传力评价和育种值预测产生了不良影响。养鹿业的持续发展不仅仅是鹿茸产量的持续提高,也包括了未来市场对质量和品种的需求,因此需要储备更多的遗传资源以适应未来环境的变化。现阶段我们需要科学、合理地评价中国的家养梅花鹿资源,制定合理的保种计划,并采取切实可行的保种措施,才能保证养鹿业的可持续发展。开发遗传标记构建分子系谱,对其群体内和个体间的遗传关系有了进一步的了解,建立系谱用于梅花鹿育种改良,配种方案的制定、血缘关系、近交率的确定,以及群体遗传参数的估计,进而提高梅花鹿群体总体质量,另一方面也可以为梅花鹿遗传资源的有效保护提供理论依据。
短串联重复序列(shorttandemrepeat,str)又称微卫星dna(microsatellitedna),是一种高度重复序列,由核心序列和侧翼序列构成。核心序列碱基数为1-6bp,其中核心序列为1-4bp的微卫星最为常见。核心序列的重复数决定了微卫星标记多态性,而侧翼序列将微卫星特异定位于染色体的某一位置。在真核生物的染色体上存在大量的微卫星位点。微卫星标记是一种中性遗传标记,不编码蛋白质和rna,但既可以分布于基因组的编码区,也可以分布非编码区。分布于非编码区的微卫星标记是最为常见的遗传标记,因为它不受选择压的影响,能够从根本上反映变异的进程,而分布于编码区的微卫星标记则有可能由于影响的蛋白质功能而受到选择的影响。str位点在基因传递过程中遵循孟德尔共显性方式遗传,因其片段短、扩增效率高、判型准确等特点,已广泛应用于法医学个体识别和亲子鉴定等领域。自1990年初以来,微卫星标记广泛适用于人口结构和父权分析,它相对廉价,相比二等位标记,如单核苷酸多态性,它的每个标记基因型会提供更多的群体遗传信息。
str位点的开发就是检测出str两侧的核苷酸序列,以此作为设计pcr扩增引物的根据,以便在不同品种或不同个体间扩增出多态性微卫星dna片段。用于微卫星标记的开发技术归纳起来主要有6种,分别是:直接文库筛选法、基于锚定pcr技术的方法、单引物延伸富集法、选择杂交富集法、ssr转移扩增法、生物信息学方法。
基因组文库筛选法是最经典的开发微卫星标记的方法,即通过构建基因组文库、筛选文库、阳性克隆测序获得微卫星序列。构建基因组文库一般是采用限制性内切酶将基因组dna消化为小片段,也可以采用超声波或喷雾器等方法,然后将小片段连入载体转入大肠杆菌,这样就构建成了基因组文库。文库的筛选通常用同位素标记的微卫星探针杂交筛选,最终通过测序获得微卫星序列,根据侧翼序列设计引物。但是筛选基因组文库工作量大,效率低,因此仅适用于基因组中微卫星含量高的物种。
基于锚定pcr技术的方法是利用5’锚定简并引物以基因组dna为模板进行扩增,再将扩增产物连入载体进行克隆测序获得微卫星标记的方法。2001年在锚定pcr法的基础上,又发明了两种开发微卫星标记的方法,sam和stmp。sam即选择扩增微卫星法(selectivelyamplifiedmicrosatellite),它是5’锚定pcr技术与选择性扩增微卫星多态性位点相结合的产物,是将两个人工接头序列连接到双酶切的dna片段两端,先后经抑制性pcr、选择性pcr和sampcr三步得到含ssr重复序列的扩增产物,再用聚丙烯酰胺凝胶分离、回收、克隆,测序后分别设计引物。stmp则是序列标签微卫星法(sequence-taggedmicrosatelliteprofiling),它是利用基因表达序列分析原理,建立富含微卫星序列的标签文库,快速大通量的分离单位点微卫星序列。该法过程较为繁琐。
单引物延伸富集法是用噬菌粒构建基因组文库,随后用辅助噬菌体超感染产生单链环状dna,以此为模板,微卫星序列作引物进行延伸反应,形成双链环状dna,然后转化大肠杆菌建成微卫星富集文库。该方法包含过多的步骤,操作复杂,而且对大肠杆菌菌种要求特殊,因此,其应用受到一定的限制。
选择杂交富集法是目前应用最为广泛的ssr分离方法,该方法操作简单,易于掌握。其原理是首先对基因组dna进行片段化处理,接着进行大小选择,回收大小在200-1000bp之间的片段,随后在回收片段两端连接一个接头分子,由于该接头含有后续引物结合序列,因此可用以扩增连有接头的dna分子及杂交后富集扩增。连有接头的dna片段随后便与固定在尼龙膜上的ssr探针进行杂交,也可以与生物素标记的ssr探针进行杂交。经杂交富集的片段洗脱后用接头序列特异引物进行扩增,连接到克隆载体转化大肠杆菌形成富集文库。
ssr转移扩增法,ssr侧翼序列在亲源关系较近的物种之间具有保守性,因此可以在近缘物种之间转移扩增。但研究认为植物ssr的种间扩增仅限于同属植物或密切相关的属间,并且这些能够转移的ssr很可能属基因内ssr,其多态性不高。
生物信息学方法是利用生物学软件从genbank、ddbj、embl等公共数据库中检索ssr序列,是一种简便快捷的方法。具体操作包括dna序列的下载、微卫星搜索软件搜索含微卫星的dna序列、设计引物、在相应物种中进行扩增。这种方法相较上述几种方法更简单方便,但是它只能应用于水稻、拟南芥等序列信息已知的物种,目前该方法主要用于est-ssr的开发。随着各种生物基因组测序计划的不断开展,生物信息学方法势必将成为一种快速、简便、实用的微卫星开发技术。
梅花鹿基因组计划的初步完成,为梅花鹿遗传标记的深度开发带来了机遇。基因组庞大规模序列的可利用性、高通量基因表达检测方法的发展及大规模数据分析能力的提高,为str位点发现展现了广阔的前景。然而,str位点的筛选不等于str位点的应用。目前,str位点的主要挑战之一,就是要快速估测和了解该位点的应用价值,确定该位点是否能够用于梅花鹿种群遗传结构评价和亲缘关系鉴定研究。str位点的筛选研究正在成为当今物种遗传资源评价的重要途径,同时也为深入了解梅花鹿现有群体的遗传背景、种群内部和种群间的亲缘关系以及品种培育、保种策略的制定奠定了基础。这一领域的迅速发展很大程度上借助于众多技术方法的发展和应用。所以,发现和鉴定梅花鹿特异的str位点,通过群体遗传学的研究、保种和育种计划的实施,将促进动物资源学与生物信息科学和高新技术产业相结合;刺激相关学科与技术领域的发展,其研究成果可直接指导和转化为实际应用,具有不可估量的社会效益和经济效益。
技术实现要素:
本发明的目的是提供基于梅花鹿全基因组开发的str分子标记及其应用。
本发明的另一目的是提供一种快速、准确和有效的梅花鹿str位点开发方法。
为了实现本发明目的,本发明采用组装质量高、注释完整的梅花鹿参考基因组为str位点开发模板,该参考基因组具有以下特征:错误率低,总长度长,完整度高。采用100个梅花鹿个体重测序数据为str位点多态性搜索数据库,该数据库具有以下特征:代表性强,覆盖度高,测序深度适中。
基于梅花鹿全基因组的str位点开发方法,所述方法包括下述步骤:
(1)两步法获取str位点:第一步采用misa.perl脚本筛选梅花鹿基因组中的str位点,程序为:perlmisa.plw_vcf_revise_contig_new_6.fa,得到文件w_vcf_revise_contig_new_6.fa.misa。编辑脚本文件4_repeat_extract_from_misa.pl,提取4碱基重复单元的str位点,脚本如下:
opengff,"$argv[0]"ordie"cannotopenfastafile,$!";
while(<gff>)
{
chomp;
@f=split;
if($f[3]=~/\(\w{4}\)/||$f[3]eqssr){
print"$_\n";
}
}
运行perl4_repeat_extract_from_misa.plw_vcf_revise_contig_new_6.fa.misaw_vcf_revise_contig_new_6.fa_4repeat.misa后得到文件w_vcf_revise_contig_new_6.fa_4repeat.misa。
第二步在str位点多态性搜索数据库中筛选阈值大于7000的含有indels的str区间,编辑脚本文件ssr_indels_doc1_doc2.pl,如下:
运行perlssr_indels_doc1_doc2.plw_vcf_revise_contig_new_6.fa_4repeat.misafinal.pass.indels.vcf7000,得到文件file1和file2。编辑脚本文件extract_file1_duplication.pl,去除file1重复的str位点,脚本如下:
运行perlextract_file1_duplication.plfile1,得到file3,即有较高多态性的str位点。
(2)批量设计str引物:
采用primer3对筛选出的str位点批量设计引物。程序文件为p3_in_w_vcf_revise_contig_new_6.fa.pl,primer3和p3_out.pl。运行程序perlp3_in_w_vcf_revise_contig_new_6.fa.plfile3,得到文件file3.p3in,运行primer3软件,得到file3.p3out,运行perlp3_out.plfile3.p3outfile3,得到file3.results文件,即批量设计的引物序列。
以参考基因组为模板,采用批量设计的引物进行e-pcr,程序文件为primer_for_e-pcr.pl,e-pcr软件和filter1.pl。运行perlprimer_for_e-pcr.plfile3.resultsprimer.txt,得到primer.txt文件。运行famap–bw_vcf_revise_contig_new_6.fa.famapw_vcf_revise_contig_new_6.fa,得到w_vcf_revise_contig_new_6.fa.famap。运行fahash–bbo1.fa.hash–w12–f3w_vcf_revise_contig_new_6.fa.famap得到bo1.fa.hash。运行re-pcr–sbo1.fa.hash–n2–g1–m50–d50-1000–0bo1.epcroutprimer.txt,得到bo1.epcrout文件。运行perlfilter1.plbo1.epcroutbo1.epcrout.filter,得到bo1.epcrout.filter文件。运行cut–f1bo1.epcrout.filter|grep–f-primer.txt>final_primer.txt,得到final_primer.txt,即最终筛选到的引物序列。
(3)聚丙烯酰胺凝胶电泳筛选多态性str位点:采用聚丙烯酰胺凝胶电泳对筛选的引物进行多态性验证。引物由上海生工生物技术公司合成。提取8个无亲缘关系的梅花鹿个体dna,以提取的梅花鹿基因组dna为模板进行pcr扩增,制备聚丙烯酰胺凝胶,对扩增产物进行电泳及银染检测,挑选电泳条带多态性高的引物进行后续的测序验证。
(4)测序法鉴定str位点真实性:将str引物扩增的pcr产物进行测序,与以参考基因组为模板的e-pcr扩增序列进行比对,通过一致性分析,确定开发的str位点的真实性。
(5)群体分析验证str位点适用性:应用筛选的str标记对梅花鹿养殖群体进行检测,对群体内总体遗传多样性进行综合评价,对群体间遗传差异进行分析。excelmicrosatellitetoolkitversion3.1计算等位基因数、多态信息含量、期望杂合度和观测杂合度。fstat2.9.3.2software用于统计近f统计量。利用genepop检测位点是否符合hardy-weinberg平衡;用马尔可夫链方法分析位点的连锁不平衡。amova分析研究组间和组内群体变异程度。群体分化的方差分析通过arliquin软件计算。群体间遗传差异与分歧由arlequinversion3.5.1.3计算。使用structurerv2.2clummp,distruct软件进行贝叶斯聚类分析。以梅花鹿养殖群体的线粒体dna群体遗传分析结果为对照,确定开发的str位点的适用性。
上述方法为一种基于梅花鹿基因组的快速、准确和有效的含4碱基重复单元的梅花鹿str位点开发方法,其中,根据实际需要可以调整str位点多态性的筛选阈值,具体步骤为:将以下程序的阈值7000调整为需要的数值:perlssr_indels_doc1_doc2.plw_vcf_revise_contig_new_6.fa_4repeat.misafinal.pass.indels.vcf7000。
本发明的优点与效益:本发明对梅花鹿str位点开发方法进行了调整与优化,使str位点开发更容易;str位点筛选方法的改进,可以快速、大批量地进行多态性str位点的查找,准确率与现有技术相比有大幅提高,大幅降低了成本。本发明开发的梅花鹿str位点含有4碱基重复单元,较常规的2-3碱基重复单元辨识度更高、更稳定,基因判型更容易;可精确定位str位点在梅花鹿基因组中的位置,且本方法同样适用于2-3碱基重复单元的str位点开发,筛选效率高、快速准确,适于大批量的str位点开发。
本发明涉及的含4碱基重复单元的梅花鹿str位点可以为梅花鹿群体遗传学、亲子鉴定、同胞分析和个体鉴别研究提供便捷有效的分子标记;也可为其他动植物分子标记的开发提供新思路。
本发明提供的基于梅花鹿全基因组开发的str分子标记,所述str分子标记为str1、str2、str6、str8、str11、str18、str19、str22、str23、str34、str36、str42、str43、str48、str49、str50、str53、str58、str69、str70、str72、str74、str75、str77、str80、str82、str89、str94、str96、str97、str98中的任一个。
用于扩增上述str分子标记的引物序列分别如seqidno:1-62所示。
本发明还提供基于梅花鹿全基因组开发的str分子标记组合,所述str分子标记组合为上述str分子标记中的任意两个或多个组合。
本发明还提供用于检测所述str分子标记的引物或试剂盒。
31个str分子标记对应的引物序列分别如seqidno:1-62所示。其中,分子标记str1对应的引物序列为seqidno:1-2,分子标记str2对应的引物序列为seqidno:3-4,分子标记str6对应的引物序列为seqidno:5-6,以此类推。
本发明31个str位点的等位基因数量(numberofallelesatdifferentlocus)见图1。
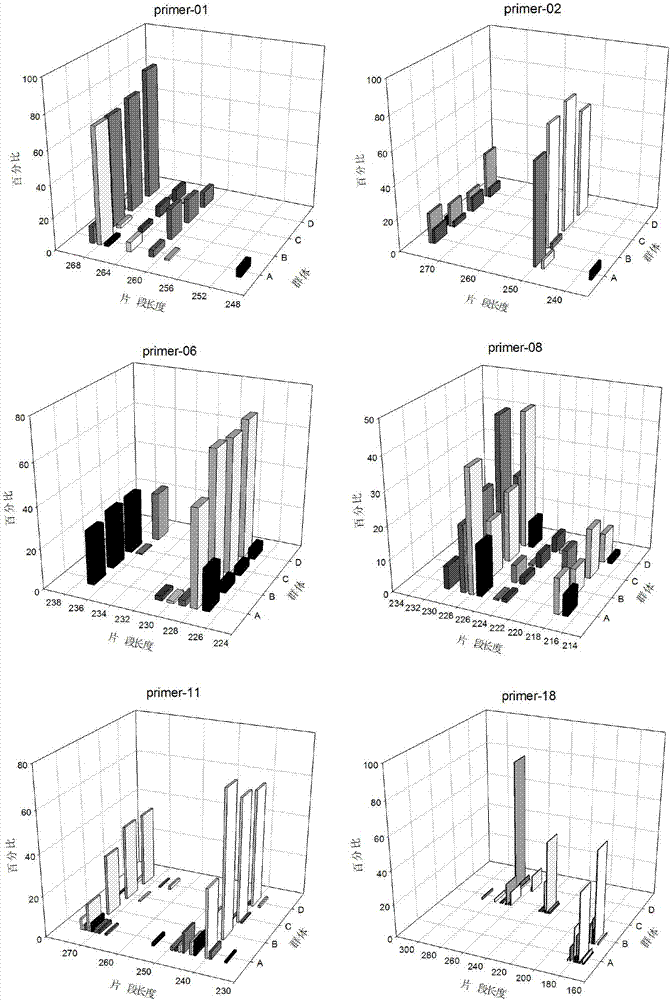
本发明31个str位点在4个群体中的等位基因频率(allelefrequenciesforallpopulationsbylocus)见图2。
本发明31个str位点的期望杂合度(expectedheterozygosities)见图3。
本发明31个str位点的多态信息含量(picvalues)见图4。
本发明根据31个str位点对4个群体构建的系统发育树(neighbour-joiningtreeshowinggeneticrelationshipsoffourstrainsofsikadeer)见图5。
本发明还提供所述str分子标记单独或组合使用在梅花鹿群体遗传学分析中的应用。
本发明还提供所述str分子标记单独或组合使用在梅花鹿亲子鉴定中的应用。
本发明还提供所述str分子标记单独或组合使用在梅花鹿品种选育中的应用。
本发明是以梅花鹿全基因组序列为基础进行str位点筛选、引物设计及应用效果验证,最终获得高扩增效率、高识别率的梅花鹿str位点标记。通过对梅花鹿str位点开发方法进行调整与优化,使str位点开发更容易;str位点筛选方法的改进,可以快速、大批量地进行多态性str位点的查找,准确率与现有技术相比有较大程度提高,大幅降低了成本。
本发明采用测序法和群体分析相结合来验证str位点的真实性和适用性。本发明开发的梅花鹿str位点含有4碱基重复单元,较常规的2-3碱基重复单元辨识度更高、更稳定,基因判型更容易;可以为梅花鹿群体遗传学研究提供便捷有效的分子标记。
附图说明
图1为本发明31个str位点的等位基因数量。
图2为本发明31个str位点在4个群体中的等位基因频率。
图3为本发明31个str位点的期望杂合度。
图4为本发明31个str位点的多态信息含量。
图5为本发明根据31个str位点对4个群体构建的系统发育树。
图6为本发明实施例4中根据31个str位点对4个群体进行的遗传结构分析。
图7为本发明实施例4中31个str位点代表的群体间遗传分歧。
图8为本发明实施例4中利用31个str位点计算的nei’s遗传距离。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。若未特别指明,实施例均按照常规实验条件,如sambrook等分子克隆实验手册(sambrookj&russelldw,molecularcloning:alaboratorymanual,2001),或按照制造厂商说明书建议的条件。
以下实施例中所用的数据库来源:
1、梅花鹿参考基因组
梅花鹿参考基因组测序以吉林省左家梅花鹿基因组一号母鹿为样本,采用二代和三代测序结合的策略,二代测序构建200bp,300bp,400bp,600bp四个插入片段文库,共测序244.6g数据,得到clean数据242.9g,覆盖基因组93x。通过kemr分析,估计梅花鹿基因组大小2.6g,contign50为13kb。三代单分子测序获得61.6gb数据,覆盖基因组23.7x,组装基因组总长接近2.5gb,达到估计基因组大小的95.4%,组装的n50contig大小为4.0mb。使用二代测序数据对三代组装结果进行纠错,使三代测序组装的错误率明显降低。估算梅花鹿基因组的杂合度为0.45%,较高的杂合度与kmer分析中估计的高杂合是一致的。cegma完整度评估显示,248个极端保守基因中completeproteins占97.58%,说明基因组完整度非常高。此外,通过bionanoirys物理图谱和梅花鹿基因组混合组装,明显提高了梅花鹿基因组的组装结果,scaffoldn50达到11mb,最终得到参考模板文件w_vcf_revise_contig_new_6.fa。
2、str位点多态性搜索数据库
对来自饲养条件基本一致的5个梅花鹿群体共100个个体进行全基因组重测序。采用百泰克溶液型全血dna提取试剂盒(百泰克dp1102)分别提取血液基因组dna,检验合格的dna样品通过covaris破碎机随机打断成长度为500bp的片段,采用truseqlibraryconstructionkit进行建库,dna片段经末端修复、加ploya尾、加测序接头、纯化和pcr扩增完成整个文库制备,制备完成后,用qubit2.0进行初步定量,稀释文库至1ng/μl,用agilent2100对文库的插入片段大小进行检测,符合预期后,使用q-pcr方法对文库的有效浓度进行准确定量,以保证文库有效浓度>2nm,库检合格的文库通过illuminahiseqxten的pe150bp模式进行测序。
对测序获得的数据进行质量过滤,使用cutadapt软件去除接头序列,使用solexaqa软件去除质量值低于20的碱基,将得到的高质量测序数据通过bwa软件比对到梅花鹿参考基因组,100个样本平均有效测序深度达到7×(梅花鹿基因组估测大小为2.6g),测序质量较高(q20=95.11%、q30=89.53%),gc分布正常,gc含量为45.38%,对基因组的覆盖度均超过93%。使用samtools进行去重复,gatk进行局部重比对,碱基质量值校正等处理,再使用gatk进行小片段插入缺失(smallindel)的检测,过滤按照条件“qd<2.0||fs>200.0||readposranksum<-20.0”对smallindel进行过滤,并得到最终的smallindel的位点集(str位点多态性搜索数据库),得到indels数量为6,545,087,数据库文件为final.pass.indels.vcf。
实施例1梅花鹿4碱基重复单元的str位点筛选和多态性初步分析
根据梅花鹿基因组测序和梅花鹿群体重测序结果,首先采用misa分析获得参考基因组中的str位点940413个,提取含4碱基重复单元的str位点14863个;然后在str位点多态性搜索数据库中筛选阈值大于7000的含有indels的str区间386个。
采用两步法可以快速在梅花鹿基因组中进行高多态性4碱基重复单元的str位点筛选。
实施例2批量设计str引物
用primer3对筛选出的str位点批量设计引物,引物长度界定在20bp±2bp,退火温度控制在55℃-60℃之间,cg%控制在30%-80%之间,剔除易产生错配和发夹结构的引物,产物片段大小控制在80-300bp之间。针对386个高长度多态性位点设计了284对str引物,用设计的引物进行e-pcr,去除非特异性扩增,通过在ncbi中进行比对分析,尽量使筛选的str标记覆盖所有染色体,避免位于染色体末端,选择100对引物进行后续的多态性验证。
采用primer3批量设计引物,对设计的引物进行e-pcr筛选验证,在ncbi数据库中进行序列比对进一步筛选,对str位点的开发方法可行。
实施例3str位点的多态性和真实性验证
提取8个无亲缘关系的梅花鹿个体dna,以提取的梅花鹿基因组dna为模板进行pcr扩增,引物由上海生工生物技术公司合成。制备聚丙烯酰胺凝胶,对扩增产物进行电泳及银染检测,根据电泳条带筛选多态性高的31对引物进行后续的测序验证。将31对引物扩增的pcr产物进行测序,与以参考基因组为模板的e-pcr扩增序列进行比对,通过一致性分析,确定31个str位点均为真实存在的多态性位点。
通过聚丙烯酰胺凝胶检测和测序分析,31个str位点均为真实存在的多态性位点。
实施例4群体分析验证str位点适用性
应用筛选的31个str标记对4个梅花鹿养殖群体共384个个体进行检测,对群体内总体遗传多样性进行综合评价,对群体间遗传差异进行分析。具体操作为:excelmicrosatellitetoolkitversion3.1计算等位基因数、多态信息含量、期望杂合度和观测杂合度。fstat2.9.3.2software用于统计近f统计量。利用genepop检测位点是否符合hardy-weinberg平衡;用马尔可夫链方法分析位点的连锁不平衡。amova分析研究组间和组内群体变异程度。群体分化的方差分析通过arliquin软件计算。群体间遗传差异与分歧由arlequinversion3.5.1.3计算。使用structurerv2.2clummp,distruct软件进行贝叶斯聚类分析。确定开发的str位点的适用性。
31个str位点在4个梅花鹿养殖群体共发现了434个等位基因,其中在位点primer18上检测到的等位基因数最多,共33个;在位点primer74和primer89等位基因为5个,是等位基因最少的位点。所选位点中多态信息含量(pic)从0.877(primer18)到0.198(primer74),平均值为0.646。从4个群体所提供的基因型数据来看,除primer43、primer74、primer80和primer97的多态信息含量较低外,其他位点的pic均≥0.5,呈现较高的多态性。另外,期望杂合度最高的是位点primer18(0.888),最低的是primer74(0.208),平均值为0.688(表1)。鉴定各位点在4个群体内哈代温伯格平衡性(表3),primer22、primer36、primer48和primer94位点在所有4个群体中都处于哈代温伯格平衡,primer08、primer18、primer19、primer70、primer72和primer82在所有4个群体中都偏离哈代温伯格平衡。
利用31对str位点的基因型代表梅花鹿基因组水平的多样性,在4个群体中,平均等位基因数分布从5.45±2.25到9.13±4.86,最高的群体是c,最低的群体是b。从观测杂合度来看,4个群体中最高的是c,最低的是a,而在期望杂合度上,最高的群体是a,最低的群体是b。该结果表明,在基因组水平上,各群体梅花鹿表现出相似的遗传多样性水平(表2)。从31对str位点表现出的群体间差异来看,4个群体间差异不大(图7),图7代表两个群体之间遗传差异大小,其绝对值越接近于1代表差异越大,越接近0代表两个群体差异越小。群体间的遗传分歧在0.092(cvsd)到0.158(avsd)之间。对于群体间遗传分歧,还用nei’s遗传距离检测了群体间差异、群体间差异平均值和群体内差异来诠释4个群体的遗传分歧和差异程度,结果也表明群体间和群体内的遗传差异不大(图8)。structure推导结果表明,4个群体最佳可能的遗传背景为k=2,即群体a和d有相同的遗传背景,群体b和c有相同的遗传背景(图6,表4)。
表131个str位点在4个梅花鹿群体内的遗传变异分析
n;等位基因数;pic:多态信息含量;ho:观测杂合度;he:期望杂合度。
表231个str位点在4个梅花鹿群体内的平均等位基因数和平均杂合度
表331个位点在4个群体内哈代温伯格平衡检验
注:*表示差异显著(p≤0.01),即偏离哈代温伯格平衡。
表4structure分析最佳k值分析表
注:*表示最佳可能的k值。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之做一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
序列表
<110>中国农业科学院特产研究所
<120>基于梅花鹿全基因组开发的str分子标记及其应用
<130>khp171114292.8
<160>62
<170>patentinversion3.3
<210>1
<211>20
<212>dna
<213>人工序列
<400>1
gcctaaaacctgtgctctgc20
<210>2
<211>20
<212>dna
<213>人工序列
<400>2
aggattcttgctttgcctga20
<210>3
<211>24
<212>dna
<213>人工序列
<400>3
cagtgaaaagaataccctgaaatg24
<210>4
<211>20
<212>dna
<213>人工序列
<400>4
aagggttgagggacataggg20
<210>5
<211>20
<212>dna
<213>人工序列
<400>5
tcttgcttcctttcttccca20
<210>6
<211>20
<212>dna
<213>人工序列
<400>6
gagggaggaaagacctcacc20
<210>7
<211>20
<212>dna
<213>人工序列
<400>7
tttgcctcagtgtcttccct20
<210>8
<211>20
<212>dna
<213>人工序列
<400>8
agctgtggttgtggtagcag20
<210>9
<211>20
<212>dna
<213>人工序列
<400>9
aaacaaccatatgcaagccc20
<210>10
<211>20
<212>dna
<213>人工序列
<400>10
ctgctgagaaagcgtgtcct20
<210>11
<211>20
<212>dna
<213>人工序列
<400>11
agtccatccctctgaagcct20
<210>12
<211>20
<212>dna
<213>人工序列
<400>12
ctctgcacaggcaatcacat20
<210>13
<211>20
<212>dna
<213>人工序列
<400>13
tgcttcctgagcagtcaaaa20
<210>14
<211>20
<212>dna
<213>人工序列
<400>14
ggatgcgtgaagagtcacaa20
<210>15
<211>20
<212>dna
<213>人工序列
<400>15
aagttcagttggtggcttgg20
<210>16
<211>20
<212>dna
<213>人工序列
<400>16
agaatgcaagggaagtggtg20
<210>17
<211>20
<212>dna
<213>人工序列
<400>17
cctttgctccatcagagctt20
<210>18
<211>20
<212>dna
<213>人工序列
<400>18
ggcaggcaaatgggttacta20
<210>19
<211>20
<212>dna
<213>人工序列
<400>19
tgcccacatctgcatttcta20
<210>20
<211>20
<212>dna
<213>人工序列
<400>20
atgggcaaggcctgataata20
<210>21
<211>20
<212>dna
<213>人工序列
<400>21
tcatttccatgtgcttgaca20
<210>22
<211>20
<212>dna
<213>人工序列
<400>22
aggtgggcaagcaaaatatg20
<210>23
<211>20
<212>dna
<213>人工序列
<400>23
agcagggagcacttttcctt20
<210>24
<211>20
<212>dna
<213>人工序列
<400>24
aacatccccaagtattgcca20
<210>25
<211>20
<212>dna
<213>人工序列
<400>25
ccgagaccgtcggtatttta20
<210>26
<211>20
<212>dna
<213>人工序列
<400>26
gtcggacgtgagtgaagtga20
<210>27
<211>20
<212>dna
<213>人工序列
<400>27
taaatgggacttccctggtg20
<210>28
<211>20
<212>dna
<213>人工序列
<400>28
gggactccctacaaacgtga20
<210>29
<211>20
<212>dna
<213>人工序列
<400>29
ttgaaggaaattttgccagg20
<210>30
<211>20
<212>dna
<213>人工序列
<400>30
ggcccagagaacttcaaatg20
<210>31
<211>20
<212>dna
<213>人工序列
<400>31
aaggaaaggggaacagagga20
<210>32
<211>20
<212>dna
<213>人工序列
<400>32
agctgcccattcatttgtct20
<210>33
<211>20
<212>dna
<213>人工序列
<400>33
cactccacacgtgaatgctt20
<210>34
<211>20
<212>dna
<213>人工序列
<400>34
tgtggaacacagtggcattt20
<210>35
<211>20
<212>dna
<213>人工序列
<400>35
attgaacctgggtctcatgc20
<210>36
<211>20
<212>dna
<213>人工序列
<400>36
ccagatccatctgagccact20
<210>37
<211>20
<212>dna
<213>人工序列
<400>37
ctgtgtgctgcaactaggga20
<210>38
<211>20
<212>dna
<213>人工序列
<400>38
ggcaaatgactgttgggtct20
<210>39
<211>20
<212>dna
<213>人工序列
<400>39
gcctgtccaaaccctacaga20
<210>40
<211>20
<212>dna
<213>人工序列
<400>40
cagctacccagggtcaatgt20
<210>41
<211>22
<212>dna
<213>人工序列
<400>41
caacattccagcataggaaaaa22
<210>42
<211>20
<212>dna
<213>人工序列
<400>42
tagctctccaggctcctctg20
<210>43
<211>20
<212>dna
<213>人工序列
<400>43
tggacagcacagatattcca20
<210>44
<211>20
<212>dna
<213>人工序列
<400>44
tgacctgctttgtttttcca20
<210>45
<211>20
<212>dna
<213>人工序列
<400>45
gctgcataatttggggctta20
<210>46
<211>20
<212>dna
<213>人工序列
<400>46
aatccctccctgaaccattc20
<210>47
<211>20
<212>dna
<213>人工序列
<400>47
aggtctcctgccctacaggt20
<210>48
<211>20
<212>dna
<213>人工序列
<400>48
tctgtcagccttctcagggt20
<210>49
<211>20
<212>dna
<213>人工序列
<400>49
tgggttcgatccctgtttag20
<210>50
<211>20
<212>dna
<213>人工序列
<400>50
tcctgaccttgtgctccttt20
<210>51
<211>20
<212>dna
<213>人工序列
<400>51
ccccaccttgaagtcagaaa20
<210>52
<211>20
<212>dna
<213>人工序列
<400>52
taaaactcggggttccactg20
<210>53
<211>20
<212>dna
<213>人工序列
<400>53
tactggggtattccaggcac20
<210>54
<211>20
<212>dna
<213>人工序列
<400>54
acaggggcagagcagagata20
<210>55
<211>20
<212>dna
<213>人工序列
<400>55
cttttcctcctggggaagtc20
<210>56
<211>20
<212>dna
<213>人工序列
<400>56
atcgcatgagccaacttctt20
<210>57
<211>20
<212>dna
<213>人工序列
<400>57
gatggagcctgtgggagata20
<210>58
<211>20
<212>dna
<213>人工序列
<400>58
tctctggtgaaggccatctt20
<210>59
<211>20
<212>dna
<213>人工序列
<400>59
tttcagctgatggcaaaatg20
<210>60
<211>20
<212>dna
<213>人工序列
<400>60
ggagggcaaagaatagcaaa20
<210>61
<211>20
<212>dna
<213>人工序列
<400>61
ggacttgcttaccaaggggt20
<210>62
<211>21
<212>dna
<213>人工序列
<400>62
cctgcagagtagaactgggaa21
- 还没有人留言评论。精彩留言会获得点赞!