一种托法替布杂质及其制备方法与流程
本发明属于药物化学领域,具体涉及一种托法替布的杂质及其制备方法。以及其作为托法替布质量控制参照标准品的用途和托法替布及其杂质的测试方法。
背景技术:
托法替布,化学名称为:3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈,cas:477600-75-2,结构式如式ii所示。
托法替布枸橼酸盐(tofacitinibcitrate),商品名xeljanz,化学名称为:3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈枸橼酸盐。cas:540737-29-9,是由pfizer公司开发的一种选择性janus激酶(jak)1/3酪氨酸蛋白激酶抑制剂,2012年11月6日,美国食品药品管理局(fda)和辉瑞公司联合宣布,枸橼酸托法替布获准用于对氨甲喋呤治疗应答不充分或不耐受的中至重度活动性类风湿关节炎(ra)成人患者。目前,日本、瑞士、等国已批准托法替布枸橼酸盐上市,主要用于治疗成人类风湿性关节。枸橼酸托法替布的结构式如式i所示。
目前,已有多篇文献资料报道了托法替布的制备和优化方法:
中国专利cn1729192:公开了一种制备方法,如图1所示,式iv在pd(oh)2/c的催化氢化条件下脱除苄基保护基,经柱层析得到式iii;iii与氰基-乙酸2,5-二氧代-吡咯烷-1-基酯发生氰基乙酰胺化反应得到式ii(即托法替布)。2009年,price,k.e.等人采用氰基乙酸乙酯作为氰基乙酰胺化反应试剂对式ii的制备工艺进行了改进。
中国专利cn101233138、cn1325498、cn102459270,公开了式i的托法替布枸橼酸盐的制备方法,由式ii和枸橼酸反应得到。
中国专利cn101233138公开了制备托法替布枸橼酸盐的另一种方法,如图2所示,式v在pd(oh)2/c的催化氢化条件下反应得到式iii;iii发生氰基乙酰胺化反应得到式ii(托法替布);ii和枸橼酸成盐即可得到托法替布枸橼酸盐。
经过对现有工艺技术的研究,本发明人参考文献cn101233138的制备方法,采用v在pd(oh)2/c的催化氢化条件下反应得到式iii;iii发生氰基乙酰胺化反应转化为式ii,最后成盐得枸盐酸托法替布。
化学反应的产物很少是具有符合药物标准的足够纯度的单一化合物。反应副产物以及该反应中所用的原材料多少也存在于产物混合物中。在api托法替布制备过程中的某些阶段,通常必须通过hplc、tlc或gc分析法分析其纯度,以测定其是否适用于继续加工和最终用在药品中。
本领域技术人员通过了解托法替布的化学结构和合成途径,发现在式iii氰基乙酰胺化生成ii过程中,双分子ii在碱性下会发生羟醛缩合反应,产生杂质式a化合物,该杂质未有cas登记号,也未见文献报道该杂质的研究及控制应用问题。
技术实现要素:
本发明所要解决的技术问题是通过对托法替布杂质的控制,提高托法替布的质量标准,为其安全用药提供重要依据。
本发明为解决上述技术问题,提供了如下技术方案:
方案1:提供一种式a所示化合物:
方案2:提供了方案1化合物a的制备方法,该方法包括如下步骤:
(1)将托法替布与有机溶剂和/或有机碱混合;
(2)控制温度在80至95℃条件下反应42-55小时;
(3)反应完成后,缓慢降至室温,反应液倾到入冰水中,充分搅拌,分液去水相;
(4)残余物加入有机溶剂溶解,然后静止析晶,抽滤得粗产物。
方案3:提供了方案1化合物a在控制托法替布质量中的用途。
方案4:提供了一种测定托法替布及其杂质化合物a的方法,该方法包括:
(1)提供一种托法替布溶液作测试样品;
(2)提供一种含有已知量和/或特性的式a合物的溶液作参照样品;
(3)用hplc测定(1)的测试样品和/或(2)的参照品,确定托法替布样品中式a的存在或/和量。
本发明提供了一种分离的托法替布杂质,即式a化合物的制备方法及作为托法替布参照标准的用途。因此,本发明有效解决了现有技术中的缺乏对单个杂质的控制不足之处,为进一步控制托法替布的质量提供了保障。
附图说明
图1托法替布的hplc图谱
图2式a化合物的hplc图谱
图3托法替布粗品中式a化合物含量的hplc图谱
具体实施方式
在一些实施方式中,提供了式a所示化合物。
在一些实施方式中,提供了一种式a化合物的制备方法,该方法包括如下步骤:
(1)将托法替布与有机溶剂和/或有机碱混合;其中有机溶剂为c1-6脂肪醇、乙酸乙酯,丙酮,乙腈,优选c1-6脂肪醇,更优选为乙醇;有机碱为吡啶、dbu、三乙胺或n,n-二异丙基乙胺,优选dbu,有机碱与托法替布的摩尔比为5~10:1,优选7.5:1;
(2)控制温度在80至95℃条件下反应42-55小时,优选为85-95℃下反应48-55小时,更优选为85℃下反应55小时;
(3)反应完成后,缓慢降至室温,反应液倾到入冰水中,充分搅拌,分液去水相;
(4)残余物加入有机溶剂溶解,然后静止析晶,抽滤得粗产物;
优选地,该方法进一步包括将粗产物重结晶的步骤,重结晶的溶剂选自c1-6脂肪醇,优选为甲醇或乙醇,更优选为乙醇;
在一些实施方式中,粗品与重结晶溶剂的质量体积比为1:10;
在一些实施方式中,提供了一种式a化合物在控制托法替布质量中的用途;
在一些实施方式中,提供了一种测定托法替布及其杂质化合物a的方法,该方法包括:
(1)提供一种托法替布样品;
(2)提供一种含有已知量和/或特性的式a化合物作参照样品;
(3)用色谱法测定托法替布样品中式a化合物存在或托法替布的量;
在一些实施方式中,色谱法优选高效液相色谱法(hplc)法。测定并确定托法替布样品中式a合物的存在或/和量的高效液相色谱法(hplc)一般包括外标法、内标法、加校正因子的主成分自身对照法等,这些方法的具体操作过程都本领域常规的知识。所述的确定包括根据托法替布样品和参照样品的测试结果计算或对比式a合物在托法替布样品中的存在和/或量。所述计算是本领域常规的知识和计算公式。在实际生产的托法替布样品中检测的式a合物的含量在常规杂质限量以下,即不大于0.1%。
在一些实施方式中,所述的确定是指根据托法替布测试样品和参照样品的测试结果采用外标法、内标法或加校正因子的主成分自身对照法计算或判断式a化合物在托法替布样品中的存在和/或量;
在一些实施方式中,hplc检测方法采用十八烷基硅烷键合硅胶为填充剂,检测波长为289nm,选择适宜流动相使测试满足常规要求,记录色谱图,用面积归一化法计算纯度。
应该理解,本领域技术人员基于此处公开的内容,可以对本发明进行各种不偏离本发明精神和范围内的各种修改和改进。它们应当都落在本申请的权利要求定义的专利保护范围内。此外,应该理解,此处提供的实施例仅用于说明本发明的目的,而不应解释为本发明的限制。
下面的实施例用于进一步理解和说明本发明,但不限制本发明的范围。
一般操作
质谱分析使用的仪器是agilent1200高效液相色谱系统、agilent公司g6410a串联三重四级杆质谱仪,离子源采用电喷雾离子源,正离子模式。分流的hplc洗脱液,允许大约为1μg/ml进入质谱仪的离子源。
本发明使用下列缩词,并具有如下含义:
实施例1
托法替布的hplc分析方法
取本品约10.0mg,精密称定,置25ml量瓶中,加流动相(乙腈-0.3%甲酸铵溶液(取甲酸铵3.0g,加水约1000ml使溶解,滤过,即得)(12:88))约10ml超声使溶解,流动相稀释至刻度,摇匀,作为供试品溶液;照高效液相色谱法(中国药典2015年版第四部通则0512)试验,用十八烷基硅烷键合硅胶为填充剂(amethystc18-h,250mm×4.6mm,5µm);以0.3%甲酸铵溶液(取甲酸铵3.0g,加水约1000ml使溶解,滤过,即得)为流动相a,乙腈为流动相b,流速为每分钟1.0ml,照下表进行梯度洗脱;检测波长为289nm;理论板数按3-羟基-3-((3r,4r)-4-甲基-3-(甲基(7h-吡咯并[2,3-d]嘧啶-4-基)氨基)哌啶-1-基)-2-(3s,4r)-4-甲基-3-(甲基(7h-吡咯并[2,3-d]嘧啶-4-基)氨基)哌啶-1-羰基)戊二腈峰计算不低于5000。取对照溶液20µl注入液相色谱仪,记录色谱图,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。再分别精密量取供试品溶液与对照溶液各20µl,注入液相色谱仪,记录色谱图。扣除溶剂空白峰和梯度洗脱峰,量取主峰面积与各杂质峰面积的和,按面积归一法计算,含量不得小于90.0%。
实施例2
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,加入正丁醇25ml,量取dbu12ml,充分搅拌,体系缓慢升温,控温至85℃,反应55h,tlc检测,原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.5g,收率:50.0%。hplc:80.5%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例3
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,量取dbu15ml,充分搅拌,体系缓慢升温,控温至85℃,反应55h,tlc检测,原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.5g,收率:50.0%。hplc:82.5%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例4
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,量取dbu18ml,充分搅拌,体系缓慢升温,控温至85℃,反应55h,tlc检测,原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩
得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.6g,收率52.0%。hplc:84.5%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例5
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,量取dbu18ml,充分搅拌,体系缓慢升温,控温至95℃,反应42h,tlc检测,原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩
得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.2g,收率:44.0%。hplc:83.6%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例6
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,量取dbu18ml,充分搅拌,体系缓慢升温,控温至85℃,反应48h,tlc检测,原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩
得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.8g,收率:56.0%。hplc:85.6%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例7
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,量取dbu18ml,充分搅拌,体系缓慢升温,控温至80℃,反应48h,tlc检测,原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩
得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.3g,收率:46.0%。hplc:82.5%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例8
室温下,取5g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入50ml单口瓶中,量取dbu24ml,充分搅拌,体系缓慢升温,控温至85℃,反应42h,tlc检测反应不完全,继续反应至48h,基本反应完全,停止反应,自然冷却至室温,将反应液倒入150ml冰水中,搅拌30min,分液去水相。将浓缩得到粘稠物用25ml乙醇溶解,静止析晶24h。减压抽滤,得到的固体杂质粗品2.7g,收率:54.0%。hplc:85.3%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例9
室温下,将10g3-[(3r,4r)-4-甲基-3-[甲基(7h-吡咯[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈加入100ml单口瓶中,量取36mldbu,充分搅拌,体系缓慢升温,加热至85℃,反应48h,tlc检测,待原料基本反应完全,停止反应,自然冷却至室温,将反应液倒入300ml冰水中,搅拌30min,分液去水相。将得到粘稠物用50ml乙醇溶解,静止析晶24h。减压抽滤,得到固体杂质粗品6.1g。收率:61.0%,hplc:85.6%,质谱[esi-ms,m/z]中m+h+峰为625。
实施例10
取2.5g杂质粗品,加入50ml圆底瓶中,量取乙醇25ml,搅拌,加热至回流,溶清后自然降温,静止析晶过夜,抽滤,得到的固体40℃减压干燥,重复上述结晶方式两次,得产品2.05g,收率:82%,hplc95.1%。[esi-ms,m/z]中m+h+峰为625。
实施例11
将得到的杂质粗品5g,加入100ml圆底瓶中,量取乙醇50ml,搅拌,加热至回流,溶清后自然降温,静止析晶过夜,抽滤,得到的固体40℃减压干燥,重复上述结晶方式三次,得产品3.9g,收率:78%,纯度:95.8%。esi-ms,m/z]中m+h+峰为625。
实施例12
托法替布、式a化合物的定性和定量分析
a.色谱条件
色谱柱&填充物:十八烷基硅烷键合硅胶为填充剂的色谱柱(waters
symmetryc18,5μm,4.6mm×250mm)
柱温:25℃
检测波长:289nm
进样量:20µl
流速:1ml/min
流动相:以0.3%甲酸铵溶液为流动相a,以乙腈为流动相b,按下表比例做线性梯度洗脱(比例为其体积比)
b.制备鉴别托法替布、式a化合物的标示物溶液
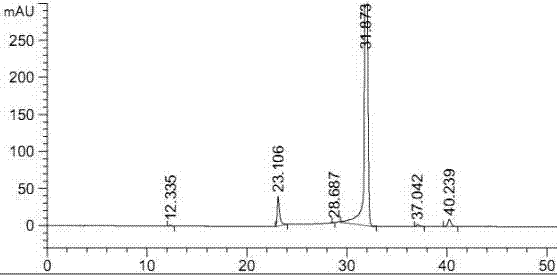
分别取托法替布,式a化合物适量,各自分别用流动相(乙腈-0.3%甲酸铵溶液(取甲酸铵3.0g,加水约1000ml使溶解,滤过,即得)(12:88))约10ml超声使溶解,流动相稀释至刻度,摇匀,即得各标示物溶液,将各标示物溶液分别注入色谱柱中测定,记录色谱图,即得各自的保留时间。在上述色谱条件下,托法替布的保留时间约为19.946min(见附图1);式a化合物的保留时间约为31.873min(见附图2),托法替布粗品中式a化合物,hplc含量0.12%,(见附图3)。依据本领域的知识,可以理解,托法替布、式a化合物各物质间均能达到有效分离,故该色谱条件能将托法替布和式a化合物准确鉴别。
- 还没有人留言评论。精彩留言会获得点赞!
- 枸橼酸托法替布及其药用组合物在制备治疗干燥综合症的药物中的应用
- N-甲基-n-((3r,4r)-4-甲基-1-苄基-3-哌啶基)-7h-吡咯并[2,3-d]嘧啶-4-胺的一种结 ...的制作方法
- 一种枸橼酸托法替布的合成方法
- 一种托法替布类似物及其制备方法与应用
- N-[(3R,4R)-1-苄基-4-甲基哌啶-3-基]-N-甲基-7H-吡咯并[2,3-d]嘧啶-4-胺晶体的制作方法
- 一种双-(3r,4r)-1-苄基-n,4-二甲基哌啶-3-胺l-二对甲基苯甲酰酒石酸盐的合成方法
- 一种适合枸橼酸托法替尼工业化的生产方法
- 托法替布中间体杂质、托法替布杂质及其合成方法,以及托法替布的质量监控方法
- 托法替布中间体的制备方法及利用其制备托法替布或其盐的方法
- 一种用液相色谱法分离测定枸橼酸托法替尼及其光学异构体的方法