一种精确定量检测食品中创伤弧菌的试剂盒及检测方法与流程
本发明属于分子生物学细菌检测领域,更具体地,涉及一种定量检测创伤弧菌的试剂盒及检测方法。
背景技术:
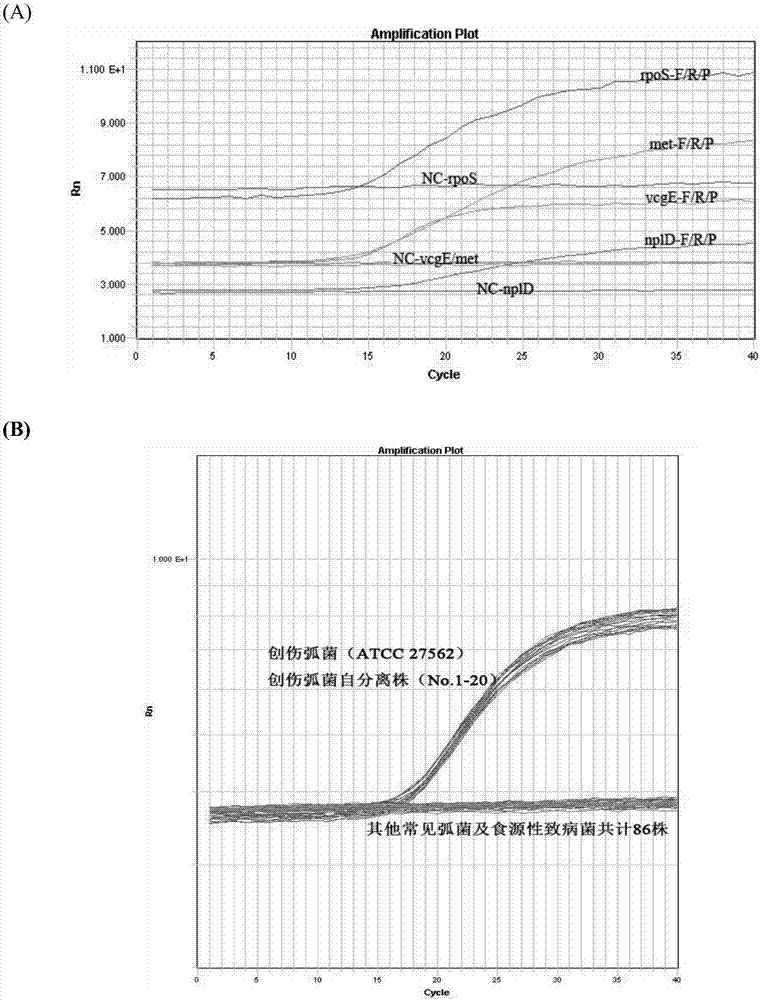
:创伤弧菌是普遍生存在海洋中的一种细菌,若生食受该菌污染的海产品(特别是牡蛎),或皮肤伤口接触存在创伤弧菌的贝类或海水,可在48小时内出现感染性休克、皮肤肌肉坏死、脓毒血症,进而引起多脏器功能衰竭,其死亡率高达60%,是美国海产品消费引起死亡的首要原因。当前评价水产品安全性的指标仍主要依赖大肠菌群等卫生指标,而这并不能反应创伤弧菌的污染程度。尽管现有法规并没有对贝类等水产中的创伤弧菌数量作出明确的规定,美国fda却支持对捕获的贝类产品进行处理,以减少或去除创伤弧菌,因此,定量方法对于准确而有效地评估水产脱菌处理措施的有效性是十分必要的,也有助于相关部门对水产安全性的监管和风险评估。目前创伤弧菌的定量检测仍以传统培养法为主,如平板计数和mpn法,往往需要经过过夜培养、选择性平板分离、生化鉴定、血清学试验等复杂的过程,不仅耗时费力,而且样品中潜在的其他杂菌有可能会因过度增殖而给目标菌鉴定造成干扰。此外,水产品中的创伤弧菌经常以“活的不可培养状态(vbnc)”存在,传统培养法无法有效检测到这部分细菌,从而影响检测结果的准确性。近年来,以实时荧光pcr(qpcr)为代表的分子生物学技术也逐渐被广泛应用到创伤弧菌的定量检测中去。相比较于传统培养法,qpcr快速,简便,灵敏、特异,但该方法属于相对定量,结果的准确性和重复性在很大程度上依赖于所绘制的标准曲线质量及扩增效率,并且需要具备已知浓度的核酸标准品。目前尚没有一个官方权威机构可以提供这种具有明确量值(即核酸拷贝数或质量浓度)的创伤弧菌核酸标准品。在实际检测中对自行构建的核酸标准分子的定量分析多采用紫外分光光度法,得到的是260nm处所有核酸分子的吸光度值,标准品中其他dna或rna分子以及蛋白等杂质成分均会影响测定结果的准确性。正是由于qpcr定量检测中缺乏统一的核酸标准品,加上样品中存在不同程度的核酸扩增抑制成分干扰导致扩增效率的差异,因此,不同实验室间qpcr的定量检测结果往往缺少可比性。新兴的微滴式数字pcr(ddpcr)被认为是第三代核酸扩增技术,通过把稀释到一定浓度的dna分子分布到10000~20000个微滴中,使大部分微滴中dna分子的数量为1或0,然后通过pcr扩增和阳性信号的累积来读取阳性反应单元数,再根据泊松分布计算出样本中的dna分子数,从而实现对dna分子的绝对定量。目前该技术用于微生物的定量分析尚处于起步阶段,相关的研究如临床样本中人疱疹病毒、耐甲氧西林的金黄色葡萄球菌、沙眼衣原体、寄生虫隐孢子虫,牛粪便样本中产志贺毒素的大肠埃希氏菌,以及导致梨火疫病和马铃薯褐腐菌病的植物病原菌等,尚未见到可用于食品中创伤弧菌定量检测的相关报道。由于食品基质多富含蛋白、脂肪、果胶等核酸扩增抑制成分,背景菌构成复杂且浓度较高,因此,ddpcr对食品中创伤弧菌定量分析的有效性和实用性尚未可知,利用ddpcr精确定量检测水产等高风险食品中创伤弧菌,可以促进风险评估工作的高效、顺利进行,具有非常广阔的应用前景。技术实现要素:本发明的目的是提供一种具有高灵敏度、高准确度和精确度、高特异性,可实现精准定量的微滴式数字pcr检测食品中创伤弧菌的试剂盒和方法,用于食品中创伤弧菌的快速、定量、精确检测。本发明的第一个目的是提供一组用于精确定量检测食品中创伤弧菌的引物和探针。本发明的第二个目的是提供一种所述引物和探针在制备食品中创伤弧菌检测试剂盒中的应用。本发明的第三个目的是提供一种食品中创伤弧菌微滴数字pcr精确定量检测的试剂盒。本发明的第四个目的是提供一种精确定量食品中创伤弧菌的检测方法。为实现上述目的,本发明采用以下技术方案:本发明分别选取创伤弧菌具有种属特异性且高度保守的单拷贝脂蛋白d基因(npld,genbankno.ay187681.1)、rna聚合酶亚单位σ因子s基因(rpos,genbankno.ay187681.1)、毒力相关基因e(vcge,genbankno.ay626581.1)和金属蛋白酶基因(met,genbankno.u50548.1)作为靶序列,通过ncbi在线工具进行序列分析和比对,利用primeexpress软件v3.0(abi,fostercity,ca,usa)设计出4对引物和探针组合,序列见表1。进一步地,在探针5’端标记fam,3’端标记bhq。引物和探针均由北京六合通经贸有限公司合成。通过qpcr和ddpcr的方法,对设计的4对引物和探针组合进行筛选和特异性验证,最终选取灵敏性和特异性最佳的met-f/r/p组合,用于后续的定量分析试验和试剂盒的组装。表1ddpcr引物和探针序列本发明提供了上述特异性引物和探针组合在制备食品中创伤弧菌检测试剂盒中的应用。本发明提供了一种食品中创伤弧菌微滴数字pcr精确定量检测的试剂盒。所述试剂盒包括所述用于检测食品中创伤弧菌的引物和探针。进一步地,所述试剂盒还包括完成数字pcr反应所需材料和试剂,例如阳性对照、阴性对照、ddpcrmastermix、微滴生成油、微滴生成卡、96孔板和铝箔热封膜等,上述材料和试剂优选为单独包装。进一步地,所述阳性对照为创伤弧菌atcc27562基因组dna,所述阴性对照为大肠埃希氏菌atcc25922基因组dna。本发明还提供了一种食品中创伤弧菌的检测方法,特别是一种利用数字pcr技术精确定量食品中创伤弧菌的方法,该方法包括:(1)提取待测样品dna,可以使用现有技术中已知的提取方法或商业化试剂盒进行提取;(2)配制ddpcr反应体系,其中,所述反应体系包括序列表中任意一组特异性引物和探针。优选地,在本发明的一个具体实施方案中,ddpcr反应体系如表2所示;表2ddpcr反应体系组分工作液浓度加样量μl反应体系终浓度ddpcrmastermix2×101×met-f10μm1.5750nmmet-r10μm1.5750nmmet-p10μm1.5750nmdna模板100~60ng/μl2/去离子水/3.5/(3)将步骤(2)配制的双重ddpcr反应体系和微滴生成油加入微滴生成卡中,置于微滴生成仪中生成微滴;(4)将生成的油包水的微滴转移至96孔板中,进行扩增反应,反应条件见表3;表3ddpcr反应条件注:升降温速度应≤2.5℃/s(5)结果判定:将扩增后的96孔板置于微滴读取仪(qx100,bio-rad,pleasanton,ca)中,利用quantasoft软件进行结果读取和分析。含有扩增产物的阳性微滴与不含扩增产物的阴性微滴会呈现出荧光信号强度的差异,以阴性微滴簇荧光幅度的最高点为界来设定阈值线。按照泊松分布原理计算获得创伤弧菌靶基因的拷贝数。本发明的有益效果如下:(1)ddpcr针对创伤弧菌单拷贝基因met进行直接绝对定量,检测灵敏度高,可以达到113拷贝(cfu)/g;(2)无需进行增菌培养及后续的特征性菌落鉴定、生化分析等步骤,大幅缩短检测时间,简化操作流程;(3)直接提取水产等样品匀浆dna,无需对样品进行复杂的稀释和检测重复,减少了稀释偏差对定值结果的影响;(4)ddpcr针对细菌基因组的检测,能有效检测到活的不可培养状态(vbnc)的细菌,从而弥补mpn和平板计数等培养法在vbnc细菌检测中的欠缺;(5)无需依赖任何核酸标准品,不需要绘制标准曲线,避免了由pcr扩增效率差异所导致的偏差;(6)由于ddpcr是终点检测,对样品中抑制剂的耐受度更高;(7)特别适于低污染水平样品的定量分析,具有更高的精确度和重复性,易于标准化。附图说明下面结合附图对本发明的具体实施方式作进一步详细的说明。图1示出qpcr对引物探针的筛选(a)与特异性验证(b)。图2示出ddpcr对引物探针的筛选(a)与特异性验证(b)。图3示出梯度稀释的创伤弧菌纯菌液qpcr扩增图谱(a)与ddpcr扩增微滴分布图(b)。图4示出创伤弧菌纯菌液中基因组浓度定值结果比较。其中x轴代表ddpcr定值检测时对纯菌液的稀释倍数,实线代表各稀释度菌液ddpcr定值结果的对数平均值(9.46),点线代表利用核酸分析仪测得结果的对数平均值(9.57),虚线代表平板计数测得的结果的对数平均值(9.11,按照1cfu菌落含1拷贝基因组计算)。图5示出人工污染牡蛎样品qpcr检测扩增曲线图(a)和人工污染牡蛎样品的ddpcr扩增微滴分布图(b)。图6示出qpcr(a)和ddpcr(b)对人工污染牡蛎样品中创伤弧菌含量的测定结果同平板计数预计值间的相关性分析。具体实施方式实施例1ddpcr引物、探针设计为实现对食品中创伤弧菌的特异性检测和绝对定量分析,我们分别选取创伤弧菌具有种属特异性且高度保守的单拷贝脂蛋白d基因(npld,genbankno.ay187681.1)、rna聚合酶亚单位σ因子s基因(rpos,genbankno.ay187681.1)、毒力相关基因e(vcge,genbankno.ay626581.1)和金属蛋白酶基因(met,genbankno.u50548.1)作为靶序列,通过ncbi在线工具进行序列分析和比对,利用primeexpress软件v3.0(abi,fostercity,ca,usa)设计出4对引物和探针组合,经过筛选最终获得特异性强、适用于ddpcr扩增条件的引物/探针组合4套,序列见表1。引物和探针均由北京六合通经贸有限公司合成。引物/探针位置分别参考genbankno.ay187681.1(npld、rpos基因)、ay626581.1(vcge基因)和u50548.1(met基因)序列信息。实施例2引物、探针的筛选与特异性验证首先,采用qpcr方法对已设计合成的引物探针进行筛选。通过对创伤弧菌(atcc27562)基因组dna的检测,发现所有引物探针均能有效扩增出靶基因,但引物探针组met-f/r/p的荧光信号出现时间最早,同阴性对照相比,其荧光信号强度最大,因而扩增效果最好(图1a)。接下来,为验证扩增反应的特异性,采用qpcr方法选取met-f/r/p引物探针,对表4中所列常见弧菌和食源性致病菌dna进行检测。本实验共使用107株菌(表4),其中包括创伤弧菌标准菌株1株、自分离株20株,其他弧菌标准/参照/自分离菌株66株,以及其他常见的食源性致病菌标准菌株20株。所有弧菌和非弧菌菌株分别在t1n1琼脂平板和bhi琼脂平板上活化3代后,挑取单菌落分别接种于5ml3%氯化钠碱性蛋白胨水和5mlbhi液体培养基中,37℃110rpm/min震荡培养18h,取1ml菌液进行10倍梯度稀释至10-9,进行细菌基因组dna的提取。结果发现创伤弧菌标准菌株(atcc27562)和20株自分离株均出现met基因阳性扩增信号,其他常见弧菌及食源性致病菌met的检测均呈阴性(图1b),表明该组引物、探针具有良好的特异性。表4实验用菌株列表注:atcc:美国典型培养物保藏中心;cmcc:医学微生物菌种保藏管理中心;cgmcc:中国一般微生物典藏中心;bjciq:北京出入境检验检疫局;gdciq:广东出入境检验检疫局。进一步地,采用ddpcr对创伤弧菌(atcc27562)基因组dna进行检测,进一步验证上述引物、探针的有效性。结果发现所有引物、探针扩增后均能出现明显的阳性微滴簇和阴性微滴簇,且引物探针组met-f/r/p的荧光信号最强(图2a),扩增效果最好。进一步利用常见弧菌对ddpcr检测体系中引物探针的特异性进行验证,发现同qpcr检测结果一样,创伤弧菌(atcc27562)出现met基因阳性微滴簇,副溶血性弧菌、溶藻弧菌、创伤弧菌、拟态弧菌与河流弧菌met基因ddpcr检测均呈阴性(图2b)。综上可见,met-f/r/p灵敏、特异,且同时满足qpcr和ddpcr的扩增条件,因此将其用于后续的定量分析试验和试剂盒的组装。实施例3试剂盒组成试剂盒包括实施例1设计的用于检测创伤弧菌的引物和探针,阳性对照(创伤弧菌atcc27562基因组dna,1000拷贝/μl),阴性对照(大肠埃希氏菌atcc25922基因组dna,1000拷贝/μl),ddpcrmastermix(购自美国biorad公司)、微滴生成油(购自美国biorad公司)、微滴生成卡(购自美国biorad公司)、twintecsemi-skirted96孔板(购自德国eppendorf公司)、铝箔热封膜(购自美国biorad公司)。实施例4ddpcr检测方法的建立1.提取样品基因组dna采用细菌基因组dna提取试剂盒(天根生化科技有限公司,货号:dp302)提取菌液dna,提取步骤按照试剂盒说明书进行,最终将核酸溶解在50μlte缓冲液中。对提取的核酸用核酸蛋白分析仪(beckmancoulter,du800)进行浓度和纯度的测定,确保提取核酸的a260/a280在1.8-2.0之间,于-20℃保存备用。2.ddpcr反应体系的配制按照表2配制20μl的ddpcr反应体系。3.微滴生成将20μl的ddpcr反应体系和70μl微滴生成油分别加入到8孔微滴生成卡中,置于微滴生成仪(qx100,bio-rad,pleasanton,ca)中生成微滴。4.扩增反应将生成的油包水的微滴(40μl)缓慢转移至96孔板中,封膜后置于pcr仪(geneamp9700,appliedbiosystems,fostercity,ca)上进行扩增反应,扩增条件如表3所示。5.结果判定将扩增后的96孔板置于微滴读取仪(qx100,bio-rad,pleasanton,ca)中,利用quantasoft软件进行结果读取和分析。含有扩增产物的阳性微滴与不含扩增产物的阴性微滴会呈现出荧光信号强度的差异,以阴性微滴簇荧光幅度的最高点为界来设定阈值线。按照泊松分布原理计算获得创伤弧菌靶基因的拷贝数。实施例5qpcr检测为便于同ddpcr方法进行比较,选用同一套引物和探针进行qpcr检测。25μl的qpcr反应体系包括:2×mastermix12.5μl,10μm的上游、下游引物各1μl,10μm的探针0.5μl,待测dna模板2μl。于abi7900荧光pcr仪(美国,abi公司)上进行扩增,50℃2min,95℃10min,接下来95℃15s、60℃1min进行45个循环。反应完成后利用sds3.2软件进行分析,根据标准曲线计算靶基因的拷贝数。实施例6创伤弧菌纯菌液的检测1.基因组dna提取与核酸蛋白分析仪测定利用3%氯化钠碱性蛋白胨水对创伤弧菌(atcc27562)的过夜培养物进行10倍梯度稀释。采用细菌基因组dna提取试剂盒(天根生化科技有限公司,货号:dp302)提取各稀释度菌液dna,提取步骤按照试剂盒说明书进行,最终将核酸溶解在50μlte缓冲液中。对提取的核酸用核酸蛋白分析仪(beckmancoulter,du800)进行浓度和纯度的测定,确保提取核酸的a260/a280在1.8-2.0之间,并按下列公式计算提取创伤弧菌基因组dna的拷贝浓度:其中m代表核酸蛋白分析仪测得的核酸浓度(ng/μl);n代表细菌基因组的长度(bp),根据ncbi上已经发布的创伤弧菌基因组的测序数据,其平均长度为4.97×106bp。创伤弧菌(atcc27562)在3%氯化钠碱性蛋白胨水中的过夜培养物1ml,提取的基因组dna(共50μl)浓度为413.6ng/μl,代入上述公式算得提取到的基因组dna浓度为7.6×107拷贝/μl,按照1拷贝基因组对应1个cfu菌落,则原始菌液浓度为3.8×109cfu/ml,其对数值为9.57。2.平板计数同时对10倍梯度稀释的创伤弧菌(atcc27562)纯菌液,分别吸取1ml菌悬液于无菌平皿内,每个稀释度做两个平皿。及时将15~20ml冷却至46℃的平板计数t1n1琼脂培养基倾注平皿,并转动平皿使其混合均匀。待琼脂凝固后,36℃倒置培养24h±2h。选取菌落数在30~300cfu之间、无蔓延菌落生长的平板计数菌落总数,每个稀释度菌落数应取两个平板的平均数。结果测得原始菌液浓度为1.3×109cfu/ml,其对数值为9.11。3.qpcr和ddpcr检测各稀释度菌液提取到的dna同时进行qpcr和ddpcr检测,结果发现ddpcr可检测到10-8稀释度(图3a),而qpcr仅检测到10-7(图3b),ddpcr的检测灵敏度高于qpcr,对低浓度菌液的检测更具优势。ddpcr在10-2稀释度达到检测上限,阳性微滴的比率达100%,自10-3至10-7稀释度菌液测得的met基因浓度均呈现出良好的线性关系(图3b,表5)。在定值范围内ddpcr测定结果具有高度一致性,由不同稀释度菌液计算出原菌液中创伤弧菌met基因含量的对数平均值为9.46,同核酸蛋白分析仪及平板计数法测得值均非常接近,分别相差了0.11和0.35个log值(图4)。实施例7人工污染牡蛎样品的检测选择经传统方法验证无创伤弧菌的牡蛎样品作为添加基质。对创伤弧菌(atcc27562)过夜培养菌悬液进行10倍梯度稀释并平板计数,同时取10-4、10-5、10-6、10-7稀释度菌液各25ml分别添加到盛有25g牡蛎样品的均质袋中,各添加浓度分别设5个样品重复,另取1份25g牡蛎样品加入含3%nacl的apw代替添加菌液作为阴性对照,上述样品用拍击式均质器拍击2min,制成系列1:1含有不同浓度创伤弧菌的污染样品。取各样品匀液2ml分别利用试剂盒(天根生化科技有限公司,货号:dp302)法提取细菌基因组dna,最终将核酸溶解在50μlte缓冲液中,进行ddpcr和qpcr检测,比较两者的定值效果。根据添加菌液的平板计数结果,估计10-7至10-4菌液污染样品中创伤弧菌含量为9.2×101~9.2×104cfu/g。在进行qpcr时,以10倍梯度稀释的创伤弧菌基因组dna(经核酸蛋白分析仪测定其原液浓度为2.91×107拷贝/μl)作为标准品,同人工污染样品中提取的dna一起进行扩增检测。结果发现4个不同污染水平的牡蛎样品均呈阳性扩增,qpcr扩增曲线如图5a所示,根据检测ct值和得到的标准曲线计算污染样品中创伤弧菌平均含量分别是415400、29102、1954、165拷贝/g(表6)。而ddpcr检测结果显示,随着污染水平的下降,出现的阳性微滴数量逐渐减少(图5b),根据阳性微滴数量和泊松分布原理,由quantasoft软件计算直接测定的创伤弧菌含量分别为133200、11610、1210、113拷贝/g(表6)。总体而言,qpcr和ddpcr的定量结果同平板计数的预计值间均存在良好的相关性(r2均大于0.99,图6),但ddpcr的定量结果更接近预计值,约为平板计数结果的1.4倍,而qpcr定量结果则为平板计数的4.5倍(图6)。ddpcr在不经增菌培养的条件下,其最低定量限即可达113拷贝/g,满足国内外对食品中创伤弧菌污染最低控制水平的要求。实施例8重复性评估上述针对创伤弧菌纯菌液和人工污染牡蛎的检测结果皆可用来评估整体检测的重复性。由表5和表6可见,在整个定量检测动态范围内,针对创伤弧菌met基因的测定结果变异系数cv普遍小于20%,满足国际上对核酸定量方法的验证要求,证明该方法用于创伤弧菌定量检测时具有良好的重复性。而且在对人工污染牡蛎样品的检测中,ddpcr的定量结果的cv普遍小于qpcr,说明ddpcr具有更好的定量重复性(表6)。表5梯度稀释的创伤弧菌纯菌液ddpcr定量检测结果表6ddpcr与qpcr对人工污染牡蛎样品中创伤弧菌的定量检测结果显然,本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定,对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。sequencelisting<110>北京出入境检验检疫局检验检疫技术中心<120>一种精确定量检测食品中创伤弧菌的试剂盒及检测方法<130><160>12<170>patentinversion3.3<210>1<211>18<212>dna<213>人工合成引物npld-f<400>1gcggtggtgactcccaaa18<210>2<211>25<212>dna<213>人工合成引物npld-r<400>2ttgcgggtttctgagtagagttatt25<210>3<211>26<212>dna<213>人工合成探针npld-p<400>3caacaccgacaacgccaaaaaaatcg26<210>4<211>25<212>dna<213>人工合成引物rpos-f<400>4gagatggagagaaggcgttattaga25<210>5<211>23<212>dna<213>人工合成引物rpos-r<400>5catgtcttcatcttgcgttgaaa23<210>6<211>28<212>dna<213>人工合成探针rpos-p<400>6tattccggatgcaaacaactctgaccca28<210>7<211>24<212>dna<213>人工合成引物vcge-f<400>7tcagaaaggctcaattgacaatga24<210>8<211>20<212>dna<213>人工合成引物vcge-r<400>8accgcctgttctgacgaaat20<210>9<211>29<212>dna<213>人工合成探针vcge-p<400>9ctcatcactgctatccaaagtagcgccaa29<210>10<211>23<212>dna<213>人工合成引物met-f<400>10ccgtattggaacaggctttatcc23<210>11<211>17<212>dna<213>人工合成引物met-r<400>11ccgcctcgaagccattt17<210>12<211>26<212>dna<213>人工合成探针met-p<400>12ttcaagctcgtagtctcgcgccagtc26当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1