酰基膦(氧)化合物及其制备方法和应用与流程
【技术领域】
本发明涉及新材料有机化学品技术领域,特别涉及一类新的酰基膦氧型结构的化合物,其化学制备工艺技术,其作为辐射固化光敏引发剂的用途,以及其在辐射固化配方产品,特别是在水性uv-led光固化涂料或油墨,等诸多场合的应用用途。
背景技术:
光引发剂化合物是一类重要的精细有机化学品材料。在以紫外光或可见(uv)光或led(即light-emittingdiode)为光源的辐射固化技术领域,可在光辐照条件下生成自由基活性物种的光引发剂化合物是诱发含烯不饱和体系进行高效光聚合反应的关键物种,因此是重要的辐射固化配方组分之一。在已经获得商业应用的众多光引发剂产品中,膦酰类化合物占据了突出的地位,其代表性产品如单酰基膦氧化合物“2,4,6-三甲基苯甲酰基二苯基氧化膦”(商品名tpo)和“2,4,6-三甲基苯甲酰基乙氧基苯基氧化膦”(tpo-l),以及双酰基膦氧化合物“双(2,4,6-三甲基苯甲酰基)苯基氧化膦”(bapo,或称irgacure819),和819的水性分散体(巴斯夫公司irgacure819-dw)。这类化合物由于兼具高引发活性,光漂白性,长波长吸收性,和uv-led广谱适用性而获得日益广泛的应用,相关的专利例如us4292154,us4737593,us6399805,ep495752等。此外,技术文献中也已存在相当数量的披露上述酰基膦氧光引发剂和其他类型光引发剂复配应用的例子,例如专利us8507726和us6777459。
在另一方面,光固化涂料和油墨产品的水性化符合了世界节能环保产业发展的战略新趋势,因而在近年来获得了日益广泛的重视和快速的发展,特别是在普适性广的水性喷涂材料,水性喷墨打印材料,和其他水性uv-led光固化材料领域,由于在施工过程中和光固化后两个阶段均完全避免了环境污染和人工健康防护难题,且led光固化技术的应用大幅度降低了能源消耗和臭氧生成,产业持续增长动力强劲。从技术上考量,水性光固化涂料或油墨的复配需要水性光固化树脂(waterborneresins)和水性光引发剂(waterbornephoto-initiators)两种关键材料;对于水性led体系固化,也需要既针对led长波发射波长(365-420纳米,特别是395-405纳米)有灵敏吸收的光引发剂,又要求该光引发剂具备良好的水溶性或水分散性。因此,设计,研发,和产业化新型的水溶性或水分散性光引发剂化合物,是当前本领域面对的关键性技术挑战问题。已知的酰基膦氧型化合物,包括上述提及的油溶性tpo,tpo-l,819,或819-dw产品,由于高度的疏水特性,无一具备足够的水溶性或水分散特性,因而难以和水性树脂体系复配形成储存稳定的涂料或油墨产品。
技术实现要素:
针对上述技术问题,本发明提供了一种新的具有较好的水溶性或水分散性,能够与水性光固化树脂有效兼容并能复配形成贮存稳定的水性光固化油墨或涂料的酰基膦(氧)化合物及其制备方法和应用。
本发明的第一个目的是提供一种通式(i)所示酰基膦(氧)化合物:
在通式(i)中:
r0是含有r3、r4、r5、r6、r7取代的芳基,或是r8、r9、r10取代的叔烷基,其中r3、r4、r5、r6、r7彼此独立的是氢、卤素原子、r、or、nrr’、ch2oh、ch2or、或ch2nrr’,其中r或r’彼此独立的选自含有1-24个碳原子的直链或支链的烷基,-c6-c12芳基,含有1-6个非连续的a元素的含有1-24个碳原子的直链或支链的烷基,含有1-6个非连续的a元素的-c6-c12芳基中的一种,其中,a元素选自氧、氮、硫元素中的一种或多种;r和r’单独存在或者连接形成一个3-6元的环系结构;r8、r9、r10彼此独立的是r,r8、r9、r10单独存在或者任意二者连接形成一个3-6元的碳环结构;
x是氧或硫,或x不存在:
n取值1-1000之间的整数;r1是r或-c(o)-r0;
当n=1时,r2-q选自氢、含有0-4个取代基的c6-c24芳基、ch2oh、ch2or、ch2oc(o)r、ch2nrr’、chr(oh)、crr’(oh)、cch3(ch2oh)oh、c(ch2oh)2oh、ch2ch2oh、ch2chmeoh、chmech2oh、ch2chphoh、ch2c(o)r、ch2co2h或其金属盐或胺盐、ch2ch2co2h或其金属盐或胺盐、ch2ch2c(o)och2ch2oh、ch2chmec(o)och2ch2oh、ch2ch2c(o)och2ch2och2ch2oh、ch2ch2c(o)oy、ch2oc(o)-nhy、ch2ch2oc(o)nhy中的一种,y是一个含有2-6个c=c双键的-c2-c36基团,
当n取值2或大于2的整数时,-r2-q是一个n元联接基团;-r2-选自-ch2-、-ch2ch2-、-ch2ch(oh)ch2-、-ch2ch(or)ch2-、-ch2ch(oc(o)r)ch2-、-ch2ch2c(o)och2ch2-、-ch2ch(me)c(o)och2ch2-、-ch2ch2c(o)och2ch2och2ch2-、-ch2ch2c(o)och2ch2ch2-、-ch2ch2c(o)och2ch2ch2ch2-中的一种;q是一个具有2个或以上-nhc(o)o-单元的小分子或聚合物基团;或者,q是一个具有2个或以上-c(o)o-单元的小分子或聚合物基团;
或者,当n取值2或大于2的整数时且当r1是r时,-r2-q是一个含有n元-ch2ch2c(o)o-、-ch2ch(me)c(o)o-、-ch2ch2c(o)c(o)o-、-ch2ch(me)c(o)c(o)o-、-ch2ch2c(o)c(o)-、-ch2ch(me)c(o)c(o)-、-ch2ch2c(o)c(o)nh-、-ch2ch(me)c(o)c(o)nh-、-ch2ch2c(o)-、-ch2ch(me)c(o)-、-ch2ch2c(o)nh-、-ch2ch(me)c(o)nh-、-ch2ch2c(o)nr-、-ch2ch(me)c(o)nr-、-ch2ch28o2-、-ch2ch(me)so2-、-ch2ch2s(o)r-、或-ch2ch(me)s(o)r-单元的小分子或聚合物基团,
或者,当n取值2或大于2的整数时,-r2-q是一个含有n元-ch2ch(oh)-、或-ch2ch(or)-单元的小分子或聚合物基团。
优选,r3=r5=r7=ch3和r4=r6=h;或r3=r7=cl和r4=r5=r6=h;或r3=r7=och3和r4=r5=r6=h;或r3=r5=r7=ch3和r4=r6=ch2oh;或r3=r5=r7=ch3和r4=r6=ch2oc(o)r;或r3=r5=r7=ch3和r4=ch2oh和r6=h;或r3=r5=r7=ch3和r4=r6=ch2or;或r3=r5=r7=ch3和r4=r6=ch2nrr’;或r3=r5=r7=ch3和r4=ch2oh和r6=ch2nrr’。
优选,r8=r9=r10=ch3;或r8=r9=r10=ch2ch3;或r8=ch3、r9-r10是环己烷结构。
优选,n取值1-100之间的整数;进一步优选,n取值1-10之间的整数。
优选,r1选自c6h5、p-ch3oc6h4、ch3、ch2ch3、ch2ch=ch2、ch2ph、c(o)cet3、c(o)cme3、联苯基、2,4,6-三甲基苯甲酰基、2,6-二甲氧基苯甲酰基、或2,6-二氯苯甲酰基中的一种。
优选,c6-c24芳基选自苯基、联苯基、萘基中的一种。
优选,y是一个含有2-6个丙烯酸酯单元的-c2-c36基团,优选,y是下列所示基团之一:
优选,当n取值2或大于2的整数时,q是下列所示基团之一:
或q是下列所示基团之一:
其中,r11是氢或r。
或优选,当n取值2或大于2的整数时且当r1是r时,q是下列所示基团之一:
或优选,当n取值2或大于2的整数时,-r2-q结构是一个含有n元-ch2ch(oh)-、或-ch2ch(or)-单元的小分子或聚合物基团,q是下列所示基团之一:
本发明的第二个目的是提供一种制备通式(i)所示酰基膦(氧)化合物的方法:自有机卤化膦r1phal2出发,制得有机膦氢r1ph2,有机膦氢r1ph2在碱作用下和酰卤r0c(o)hal或酸酐r0c(o)oc(o)r0反应,得到形式为a或b的中间体,该中间体和甲醛和氧化剂作用下,得到通式(i)所示酰基膦(氧)化合物,其中,hal为卤素;或者,中间体a或b与通式c所代表的物质在氧化剂参与下进行第一反应,得到通式(i)所示酰基膦(氧)化合物;其中,q代表一个q化学特质描述的结构,为一个含有多元环氧、多元异氰酸酯、多元不饱和羰化物、多元羧酸、多元酰卤、或多元酸酐官能基团的小分子化合物或聚合物树脂化合物;碱为无机金属氢氧化物或烷氧化物或碳酸盐或羧酸盐化合物,或有机叔胺型化合物;氧化剂选自氧气、空气、空气-金属盐复合物、金属次氯酸盐、氧气-金属盐复合物、双氧水、形式为rooh的过氧化物、过氧酸、或硫中的一种;q取值为1-n之间的整数;通式c所代表的物质选自多元环氧c1、多元异氰酸酯c2、多元不饱和羰化物c3、多元羧酸c4、多元酰卤c5、多元酸酐c6中的一种;
或者,中间体a或b和标记为r-hal的烷基卤化物或烷基拟卤素化合物发生,在第二碱存在下,亲核取代反应,制备通式(i)所示酰基膦(氧)化合物,
或,经由一个起始的膦酰原料d,在酸和甲醛共同作用下发生blanc反应,得到卤化物中间体e,其中,r取值1或2,所述酸是路易斯或质子酸,hal代表卤素原子;中间体e在亲核试剂hnu作用下发生亲核取代反应,制备得到通式(i)所示酰基膦(氧)化合物,
或,双酰基膦氧化合物(bapo)在酸和亲电试剂e+作用下,发生单膦酰基团对e+的加成反应,制备得到通式(i)所示酰基膦(氧)化合物;所述酸是路易斯酸或质子酸;亲电试剂e+是c1-c48的支链或直链的脂肪族或芳香族醛或酮,或α,β-不饱和醛、酮、酯、腈、或酰胺,或硝基烯,或烯基砜或亚砜,
或,结构f膦酰中间体sodiumbis(2,4,6-trimethylbenzoyl)phosphanide或结构gphosphinediylbis(mesitylmethanone),或者它们烯醇式的共振式异构体与与甲醛或c试剂发生反应并经过氧化剂作用,得到通式(i)所示酰基膦(氧)化合物;c试剂选自多元环氧c1、多元异氰酸酯c2、多元不饱和羰化物c3、多元羧酸c4、多元酰卤c5、多元酸酐c6中的一种,
优选,第一反应选自环氧开环反应、异氰酸酯加成反应、迈克尔共轭加成反应、酯化反应中的一种。
优选,第二碱为无机金属氢氧化物或烷氧化物或碳酸盐或羧酸盐化合物,或有机叔胺型化合物。
优选,所述酸选自三氯化铝、氯化锌、三氯化铁、三溴化铝、盐酸、氢溴酸、醋酸中的一种或多种;进一步优选,所述酸是三氯化铝或盐酸。
优选,所述hal是氯。
优选,所述亲核试剂hnu是含有氧、氮、硫、或磷原子的物质,进一步优选,所述hnu是水、nr3、hnrr’、h2nr、hsr、rssr’、hp(o)rr’、或含有氧、氮、硫、或磷原子的杂环。
优选,所述亲电试剂e+是甲醛、丙烯酸、丙烯醛、或一元或多元的丙烯酸酯单体或丙烯酸酯聚合物树脂;进一步优选,所述酸是三氯化铝或盐酸,所述亲电试剂e+是甲醛。
本发明的第三个目的是提供一种通式(i)所示酰基膦(氧)化合物在光固化配方体系中作为光引发剂或其他功能性添加剂成分的用途;或在化学合成中作为中间体或原料或试剂的用途。
本发明的第四个目的是提供一种含有上述通式(i)化合物的,可经由光辐射固化的混合物。
优选,混合物含有至少一种通式(i)化合物作为光引发剂或光引发剂组分之一;及含有至少一种含烯键不饱和化合物。
优选,以含烯键不饱和化合物总量每100份重量计算,含有的通式(i)化合物的量是0.01-30重量份;优选,以含烯键不饱和化合物总量每100份重量计算,含有的通式(i)化合物的量是0.5-10重量份。
优选,含烯键不饱和化合物是通过双键的自由基聚合反应被交联的化合物或混合物,所述含烯键不饱和化合物是单体、低聚物或预聚物、或它们的混合物或共聚物,或是他们的水性分散体。
本发明的第五个目的是提供上述混合物用作光固化涂料或油墨。
优选,上述光固化涂料或油墨在喷墨打印,纸张印刷,粘合剂,木器涂装,塑胶涂装,汽车涂装,包装材料,显示技术,建筑材料,柔性电子或光伏材料领域的应用。
本发明已经出人意料地发现,经由本发明合成方法得到的含有适当极性官能基团的酰基膦(氧)化合物,具备合适的水溶性或水分散性。我们进一步证实,这杆的新型水性光引发剂能够和水性光固化树脂有效兼容并复配形成贮存稳定的水性光固化油墨或涂料,特别是,这样的水性光引发剂首次使得水性节能环保led光固化技术得以实现,这样的技术对木器绿色涂装,环保印刷,喷墨打印,节能材料等领域拥有深远的影响。
【附图说明】
图1为本发明实施例一制备的化合物的uv-vis吸收光谱(紫外吸收光谱)图;
图2为本发明实施例十三制备的化合物的uv-vis吸收光谱(紫外吸收光谱)图。
【具体实施方式】
本项申请披露如下通式(i)所示的新型酰基膦(氧)化合物:
在上述通式(i)化合物结构中:
r0是含有r3、r4、r5、r6、r7取代的芳基,或是r8、r9、r10取代的叔烷基(如图所示),其中r3、r4、r5、r6、r7彼此独立的是氢、卤素原子、r、or、nrr’、ch2oh、ch2or、或ch2nrr’,其中r或r’彼此独立的是含有1-24个碳原子(标记为-c1-c24,下同)的直链或支链的烷基或-c6-c12芳基,r或r’结构中可以含有1-6个非连续的氧,氮,或硫元素,r和r’同时存在时其间也可以形成一个3-6元的环系结构。r8,r9,r10彼此独立的是r,三者之中的任意二者也可以形成一个3-6元的碳环结构。优选的,r3=r5=r7=ch3和r4=r6=h,或r3=r7=cl和r4=r5=r6=h,或r3=r7=och3和r4=r5=r6=h,或r3=r5=r7=ch3和r4=r6=ch2oh,或r3=r5=r7=ch3和r4=r6=ch2oc(o)r,或r3=r5=r7=ch3和r4=ch2oh和r6=h,或r3=r5=r7=ch3和r4=r6=ch2or,或r3=r5=r7=ch3和r4=r6=ch2nrr’,或r3=r5=r7=ch3和r4=ch2oh和r6=ch2nrr’。优选的,r8=r9=r10=ch3,或r8=r9=r10=ch2ch3,或r8=ch3和r9-r10是环己烷结构:
x是氧或硫,或x不存在(即式i化合物是三价膦烷);
n取值1-1000之间的整数,优选的,n取值1-100之间的整数,更优选的,n取值1-10之间的整数;
r1是r或-c(o)-r0,优选的r1是c6h5,p-ch3oc6h4,ch3,ch2ch3,ch2ch=ch2,ch2ph,c(o)cet3,c(o)cme3,联苯基,2,4,6-三甲基苯甲酰基,2,6-二甲氧基苯甲酰基,或2,6-二氯苯甲酰基;
当n=1时,r2-q结构是氢,含有0-4个取代基的c6-c24芳基(优选的是苯基,联苯基,萘基),ch2oh,ch2or,ch2oc(o)r,ch2nrr’,chr(oh),crr’(oh),cch3(ch2oh)oh,c(ch2oh)2oh,ch2ch2oh,ch2chmeoh,chmech2oh,ch2chphoh,ch2c(o)r,ch2co2h或其金属盐或胺盐,ch2ch2co2h或其金属盐或胺盐,ch2ch2c(o)och2ch2oh,ch2chmec(o)och2ch2oh,ch2ch2c(o)och2ch2och2ch2oh,ch2ch2c(o)oy,ch2oc(o)-nhy,或ch2ch2oc(o)nhy,这里y是一个含有2-6个c=c双键的-c2-c36基团,优选地y是一个含有2-6个丙烯酸酯单元的-c2-c36基团,更优选的y是下列所示基团之一:
当n取值2或大于2的整数时,-r2-q结构是一个n元联接基团;-r2-优选的是-ch2-,-ch2ch2-,-ch2ch(oh)ch2-,-ch2ch(or)ch2-,-ch2ch(oc(o)r)ch2-,-ch2ch2c(o)och2ch2-,-ch2ch(me)c(o)och2ch2-,-ch2ch2c(o)och2ch2och2ch2-,-ch2ch2c(o)och2ch2ch2-,-ch2ch2c(o)och2ch2ch2ch2-,而q是一个具有2个或以上-nhc(o)o-单元的小分子或聚合物基团,而q优选的是下列基团:
或者,q是一个具有2个或以上-c(o)o-单元的小分子或聚合物基团,而q优选的是下列基团(其中r11是氢或r):
很显然的,对于本专业从业技术人员而言,容易看出,上述q结构中的多元异氰酸酯【-nhc(o)o-】单元或多元酯【-c(o)o-】单元均源自r2结构中的羟基oh与相应的多元聚氨酯nco基团或多元酸或多元酸酐或多元酰卤进行酯化反应得来,因此上述优选q结构仅为示例性而非限定性结构,凡是符合上述化学反应实质要义的q结构均属于本发明披露范围。
或者,当n取值2或大于2的整数时且当r1是r时,-r2-q结构是一个含有n元-ch2ch2c(o)o-,-ch2ch(me)c(o)o-,-ch2ch2c(o)c(o)o-,-ch2ch(me)c(o)c(o)o-,-ch2ch2c(o)c(o)-,-ch2ch(me)c(o)c(o)-,-ch2ch2c(o)c(o)nh-,-ch2ch(me)c(o)c(o)nh-,-ch2ch2c(o)-,-ch2ch(me)c(o)-,-ch2ch2c(o)nh-,-ch2ch(me)c(o)nh-,-ch2ch2c(o)nr-,-ch2ch(me)c(o)nr-,-ch2ch2so2-,-ch2ch(me)so2-,-ch2ch2s(o)r-,或-ch2ch(me)s(o)r-单元的小分子或聚合物基团,优选的,q是下列基团:
很显然的,对于本专业从业技术人员而言,容易看出,上述q结构中的-ch2ch2c(o)o-,-ch2ch(me)c(o)o-,-ch2ch2c(o)c(o)o-,-ch2ch(me)c(o)c(o)o-,-ch2ch2c(o)c(o)-,-ch2ch(me)c(o)c(o)-,-ch2ch2c(o)c(o)nh-,-ch2ch(me)c(o)c(o)nh-,-ch2ch2c(o)-,-ch2ch(me)c(o)-,-ch2ch2c(o)nh-,-ch2ch(me)c(o)nh-,-ch2ch2c(o)nr-,-ch2ch(me)c(o)nr-,-ch2ch2so2-,-ch2ch(me)so2-,-ch2ch2s(o)r-,或-ch2ch(me)s(o)r-单元均源自如下化学结构之一的亲核(nucleophilic)特性的中间体(其中metal代表一个金属阳离子或硅烷基团,优选的metal是钠,钾,锂,sir3等)对相应不饱和酯,不饱和酰胺,不饱和砜或亚砜等的迈克尔加成反应制备。因此上述优选-r2-q结构仅为示例性而非限定性结构,凡是符合上述化学反应实质要义的小分子或聚合物型-r2-q结构均属于本发明披露范围。
或者,当n取值2或大于2的整数时,-r2-q结构是一个含有n元-ch2ch(oh)-,或-ch2ch(or)-单元的小分子或聚合物基团,优选的,q是下列基团:
很显然的,对于本专业从业技术人员而言,容易看出,上述q结构中的-ch2ch(oh)-或-ch2ch(or)-单元均源自如下化学结构之一的亲核(nucleophilic)特性的中间体对相应环氧化合物的开环反应制备。因此上述优选-r2-q结构仅为示例性而非限定性结构,凡是符合上述化学反应实质要义的小分子或聚合物型-r2-q结构均属于本发明披露范围。
通式(i)化合物的普适性制备方法之一如下面方程式所示,自有机卤化膦r1phal2(hal为卤素)出发,经由已知文献方法【cn201510020417.9】可以原位制备或分离纯化得到有机膦氢r1ph2,有机膦氢r1ph2在适当的碱base作用下和当量的酰卤r0c(o)hal或酸酐r0c(o)oc(o)r0反应,即可得到形式为a或b的中间体,该中间体和甲醛和氧化剂【o】作用下,即得到通式部分通式(i)产物;
或者,中间体a或b与通式c所代表的多元环氧c1,多元异氰酸酯c2,多元不饱和羰化物c3,多元羧酸c4,多元酰卤c5,多元酸酐c6等在氧化剂【o】参与下进行相应的环氧开环反应,异氰酸酯加成反应(酯化反应),迈克尔共轭加成反应,酯化反应等,得到另一部分通式(i)产物:这里网圈q代表一个q化学特质描述的结构,即一个含有多元环氧,多元异氰酸酯,多元不饱和羰化物,多元羧酸,多元酰卤,或多元酸酐官能基团的小分子化合物或聚合物树脂化合物。
氧化剂【o】代表氧气,空气,空气-金属盐复合物,金属次氯酸盐(优选的是次氯酸钠),氧气-金属盐复合物,双氧水,形式为rooh的过氧化物,过氧酸,或硫;q取值为1-n之间的整数。
同时,鉴于中间体a或b的亲核(nucleophilic)物种化学特质,相关联的,a或b可以和一系列标记为r-hal的烷基卤化物(氯化物,溴化物,或碘化物)或烷基拟卤素化合物(磺酸根等)发生,优选的,在合适的碱存在下,亲核取代反应,制备一部分通式(i)目标化合物。碱base是无机金属氧氧化物或烷氧化物或碳酸盐或羧酸盐化合物,或有机叔胺型化合物。
上述通式(i)化合物的制备方法我们将结合实施例子做进一步的示例性说明。
满足上述通式(i)的另一部分化合物的制备方法是,经由一个起始的膦酰原料d,在酸和甲醛共同作用下发生所谓的blanc反应,得到相应的卤化物中间体e(其中r取值1或2),这里酸是路易斯或质子酸,优选的酸是三氯化铝,氯化锌,三氯化铁,三溴化铝,盐酸,氧溴酸,醋酸,或上述酸的适当混合物;hal代表卤素原子,优选的hal是氯。关键中间体e在合适的亲核试剂hnu作用下发生亲核取代反应,即制备得到目标化合物(i)。优选的亲核试剂hnu是含有氧,氮,硫,或磷原子的物种,特别优选的hnu是水,nr3,hnrr’,h2nr,hsr,rssr’,hp(o)rr’,或含有氧,氮,硫,或磷原子的杂环。上述化学制备新技术在我们前期的发明专利申请【cn103172770】中已经有详细的披露,本发明的新的意外的有益发现是,通过选用适当的hnu试剂,利用上述技术制备出了新的具有良好水分散性或水溶解性能的酰基膦氧化合物。
制备部分通式(i)化合物的另一个方法是,如下面方程式所示,让双酰基膦氧化合物(bisacylphosphineoxide,简称bapo)例如市售的bapo-819在酸和亲电试剂e+作用下,发生单膦酰基团对e+的加成反应,从而方便地制备下述产物之一或两者同时联产。这里酸的定义与上相同,亲电试剂e+是c1-c48的支链或直链的脂肪族或芳香族醛或酮,或α,β-不饱和醛,酮,酯,腈,或酰胺,或硝基烯,或烯基砜或亚砜;优选的亲电试剂e+是甲醛,丙烯酸,丙烯醛,或一元或多元的丙烯酸酯单体或丙烯酸酯聚合物树脂;特别优选的酸是三氯化铝或盐酸,特别优选的亲电试剂e+是甲醛,在“三氯化铝或盐酸-甲醛”试剂作用下,此时下述产物中的e=ch2oh或ch2cl。
制备部分通式(i)化合物的另一个方法是,如下面方程式所示,根据coenradusjohanneshendrikushendriksen的eth-zürich博上论文中第124页记载的方法合成中间体sodiunbis(2,4,6-trimethylbenzoyl)phosphanide(即结构f)或phosphinediylbis(mesitylmethanone)(即结构g),或者它们烯醇式的的共振式异构体。然后,与前述中间体a或b类似,f或g与甲醛或系列c试剂(如前所述的c1-c6)发生反应并经过氧化剂【o】作用(氧化剂的定义与前述相同),得到目标化合物。
符合通式(i)结构的示例性化合物列举如下:
本发明进一步披露一种含有上述通式(i)化合物的,可经由光(紫外或可见光或led光或等价光源)辐射固化的混合物,特别是水性光固化配方混合物体系,以及这类配方材料体系在喷墨打印,纸张印刷,粘合剂,木器涂装,塑胶涂装,汽车涂装,包装材料,柔性电子,光伏材料等领域的应用。
这类光辐射固化配方体系的特征是:
(1)含有至少一种通式(i)所描述的化合物作为光引发剂或光引发剂组分之一;
(2)含有至少一种含烯键(c=c)不饱和化合物。
以体系中含烯键不饱和组分总量每100份重量计算,含有的通式(i)化合物的合适的量是0.01-30重量份,优选0.5-10重量份。本项申请披露的合适辐射固化体系包含的可聚合的含烯键不饱和组分是可以通过该双键的自由基聚合反应被交联的化合物或混合物,这种含烯键不饱和组分可以是单体,低聚物或预聚物,或是它们的混合物或共聚物,或是上述组分的水性分散体。
上述所述的合适辐射固化体系均可以含有根据实际需要所添加的无机或有机填充剂和/或着色剂(例如颜料或染料等),以及其它添加剂(例如紫外线吸收剂,光稳定剂,阻燃剂,流平剂,或消泡剂等)和溶剂等任意成分。
合适的自由基聚合的单体是例如含烯键可聚合单体,包括但不限于(甲基)丙烯酸酯,丙烯醛,烯烃,共轭双烯烃,苯乙烯,马来酸酐,富马酸酐,乙酸乙烯酯,乙烯基吡咯烷酮,乙烯基咪唑,(甲基)丙烯酸,(甲基)丙烯酸衍生物例如(甲基)丙烯酰胺,乙烯基卤化物,亚乙烯基卤化物等。
合适的含烯键预聚物和低聚物包括但不限于(甲基)丙烯酰官能基的(甲基)丙烯酸共聚物,聚氨酯甲酸酯(甲基)丙烯酸酯,聚酯(甲基)丙烯酸酯,不饱和聚酯,聚醚(甲基)丙烯酸酯,硅氧烷(甲基)丙烯酸酯,环氧树脂(甲基)丙烯酸酯等,以及上述物质的水溶性或水分散性的类似物。
上述无论是含烯单体还是低聚物,预聚物,或共聚物,对本专业从业技术人员而言,都是熟知的,并无特别限定。
对于本发明的要旨,我们将结合下述系列实施例进一步说明。
实施例零:2-(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)aceticacid
在氮气保护下,在室温下向含有苯基膦(12.5克,114毫摩尔)的甲苯-乙二醇二甲醚(300毫升,1/1体积比)溶液中加入叔丁醇钠(13.8克)后,再于2小时内缓缓滴加加入2,4,6-均三甲苯酰氯(24.8克),继续搅拌半小时后缓缓加入16.2克氯乙酸,加完后在室温搅拌过夜。用0.5n盐酸调节反应体系ph~2-3后缓慢滴加30%的过氧化氢水溶液(17克),加完后在40度左右反应1小时,然后往反应体系中加入100毫升水,搅拌,分层,有机相用80毫升饱合的碳酸氢钠水溶液混合80毫升水洗两次,稀盐水洗一次,硫酸钠干燥,60度下减压蒸除溶剂,残留物加入100毫升正庚烷打桨,过滤得24.7克淡黄色的固体,取部分此固体在硅胶柱色谱上用乙酸乙酯-己烷梯度洗脱得到分析纯样品。hrms(m+h)forc18h20o4p:331.1099(calculated),331.1017(experimental);(m+na)forc18h19nao4p:353.0919(calculated),353.0922(experimental)。
实施例一:((hydroxymethyl)(phenyl)phosphoryl)(mesityl)methanone
在氮气保护下,在室温下向含有苯基膦(114毫摩尔)的甲苯(300毫升)溶液中加入叔丁醇钠(13.8克)后,再于2小时内缓缓滴加加入2,4,6-均三甲苯酰氯(24.8克),加毕反应在冷水浴冷却下在30分钟内滴加浓硫酸(5.7克),加完后搅拌30分钟后,滴加37%多聚甲醛水溶液(11.0克),加完后在室温搅拌3小时。缓慢滴加30%的过氧化氢水溶液(17克),加完后在40度左右反应1小时,然后往反应体系中加入50毫升水,搅拌,分层,有机相用20毫升饱合的碳酸氧钠水溶液混合40毫升水洗两次,稀盐水洗一次,硫酸钠干燥,60度下减压蒸除溶剂,加入70毫升正庚烷打桨,过滤得26.5克淡黄色的固体粉末产品。1h-nmr(cdcl3):δ=7.87-7.84(m,2h),7.60-7.57(m,1h),7.52-7.48(m,2h),6.75(s,2h),5.55(br,1h),4.55(dd,1h),4.10(dd,1h),2.24(s,3h),2.05(s,6h);13c-nmr(cdcl3):δ=>220(c=osignal),140.7,135.7,135.4,135.2,132.7,131.4,128.9,128.8,126.9,126.3,608(d),21.1,19.4ppm;31p-nmr(cdcl3):δ=23.5ppm。hrms(m+h)forc17h20o3p:303.1150(calculated),303.1140(experimental);(m+na)forc17h19nao3p:325.0970(calculated),325.0948(experimental)。
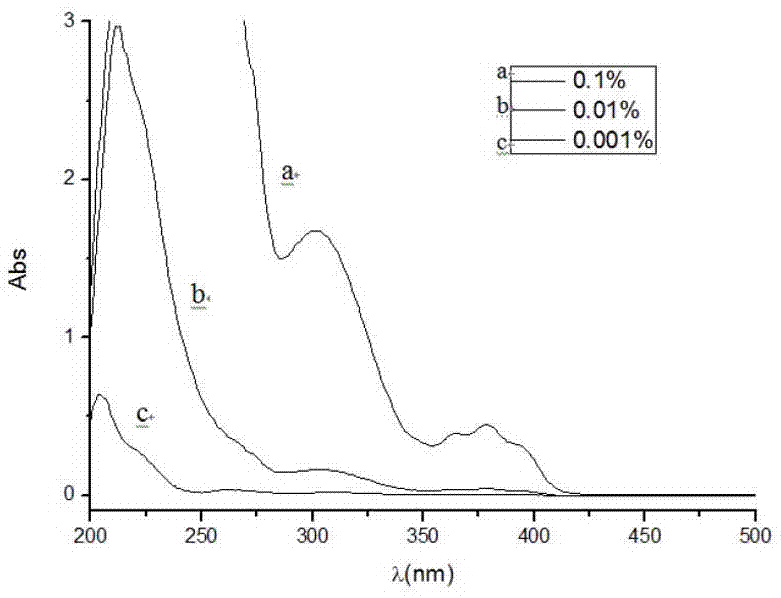
该化合物的uv-vis吸收光谱图如图1所示(溶剂为乙腈,质量百分浓度),从图可以看出该化合物在uv-led发射波长区域均有显著吸收:
实施例二:(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)methyl2,4,6-trimethyl-benzoate
在上述实施例一的合成中,同时分离得到了该实施例二化合物。1h-nmr(cdcl3):δ=7.96-7.92(m,2h),7.62-7.60(m,1h),7.55-7.27(m,2h),6.81(s,2h),6.78(s,2h),5.15(d,2h),2.27(s,3h),2.24(s,3h),2.20(s,6h),2.02(s,6h);13c-nmr(cdcl3):δ=>220(c=osignal),139.7,135.7,135.3,133.1,131.6,131.5,129.1,128.9,128.3ppm;31p-nmr(cdcl3):δ=18.1ppm。hrms(m+h)forc27h30o4p:449.1882(calculated),449.1890(experimental);(m+na)forc27h29nao4p:471.1701(calculated),471.1714(experimental)。
实施例三:((chloromethyl)(phenyl)phosphoryl)(mesityl)methanone
在氮气保护下,将418毫克irgacure819光引发剂,142毫克无水三氯化铝,72毫克多聚甲醛置于10毫升氯仿中,体系加热到75摄氏度反应45分钟,将混合物倒入冰水中,分液,干燥,过滤,浓缩后残余物在硅胶柱色谱上用乙酸乙酯梯度洗脱,得到112毫克实施例一化合物以及92毫克的本实施例目标产物。1h-nmr(cdcl3):δ=7.92-7.88(m,2h),7.62-7.60(m,1h),7.54-7.51(m,2h),6.82(s,2h),4.98(dd,1h),4.81(dd,1h),2.26(s,3h),2.10(s,6h);hrms(m+h)forc17h19clo2p:321.0811(calculated),321.0809(experiental);(m+na)forc17h18naclo2p:343.0631(calculated),343.0644(experimental)。
实施例四:((2-hydroxyethyl)(phenyl)phosphoryl)(mesityl)methanone
在氮气保护下,在室温下向含有苯基膦(2.36毫摩尔)的甲苯(20毫升)溶液中加入叔丁醇钠(464毫克)后,再于40分钟内缓缓滴加加入2,4,6-均三甲苯酰氯(431毫克),加毕反应体系继续搅拌2小时后转入一个耐压封管(sealedtube),向封管中通入环氧乙烷后继续搅拌2小时,反应液用0.86毫升的28%浓度双氧水在用硫酸调节到ph=2-3条件下处理1小时。有机相依次用1%碳酸氢钠水溶液和饱和食盐水洗涤,分液,硫酸钠干燥,过滤,得到的清液浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到402毫克淡黄色粘稠腊状的目标化合物。hrms(m+h)forci8h22o3p:317.1307(calculated),317.1326(experimental);(m+na)forc18h21nao3p:339.1126(calculated),339.1141(experimental)。
实施例五:3-(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)propanoicacid
在氮气保护下,在室温下向含有苯基膦(2.19毫摩尔)的甲苯(20毫升)溶液中加入叔丁醇钠(214毫克)后,再于40分钟内缓缓滴加加入2,4,6-均三甲苯酰氯(400毫克),加毕反应体系继续搅拌2小时后,加入丙烯酸(205毫克),反应体系转入一个耐压封管(sealedtube)在60-70度加热继续搅拌1小时。反应液用0.52毫升的28%浓度双氧水在用硫酸调节到ph=2-3条件下处理1小时。有机相依次用1%氯化铵水溶液和饱和食盐水洗涤,分液,硫酸钠干燥,过滤,得到的清液浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到286毫克淡黄色固体粉末目标化合物。hrms(m+h)forc19h22o4p:345.1256(calculated),345.1277(experimental);(m+na)forc19h21nao4p:367.1075(calculated),367.1082(experimental)。
实施例六:
在氮气保护下将160毫克的实施例一化合物和222毫克的双异氰酸酯ipdi置于8毫升的干燥甲苯溶剂中,向体系中加入6毫克的二月桂酸二丁基锡催化剂后在80度加热反应1小时,反应体系浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到334毫克淡黄色粘稠腊状目标化合物。hrms(m+h)forc46h57n2o8p2:827.3590(calculated),827.358(experimental);(m+na)forc46h56n2o8nap2:849.3410(calculated),849.3423(experimental)。
实施例七:tris((phenyl(2,4,6-trimethylbenzoyl)phosphoryl)methyl)benzene-1,3,5-tricarboxylate
在氮气保护下,将302毫克的实施例一化合物,88毫克的均三苯甲酰氯,138毫升的三乙胺,和12毫克4-二甲胺基吡啶(dmap)置于25毫升干燥的二氯乙烷溶剂中,室温搅拌反应过夜。混合物用等体积半饱和食盐水洗涤3次,有机相硫酸钠干燥,过滤,浓缩除去溶剂后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到267毫克淡黄色固体目标化合物。hrms(m+h)forc60h58o12p3:1063.3141(calculated),1063.3166(experimental);(m+na)forc60h57o12nap3:1085.2961(calculated),1085.2961(experimental)。
实施例八:2-((acryloyloxy)methyl)-2-ethylpropane-1,3-diylbis(3-(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)propanoate)
在氮气保护下,在室温下向含有苯基膦(2.36毫摩尔)的甲苯(20毫升)溶液中加入叔丁醇钠(464毫克)后,再于40分钟内缓缓滴加加入2,4,6-均三甲苯酰氯(431毫克),加毕反应体系继续搅拌2小时后,缓缓加入sartomersr351单体219毫克,此后向反应体系滴加加入0.90毫升的28%浓度双氧水(预先用硫酸调节到ph=2-3),继续搅拌反应1小时后有机相依次用1%碳酸氢钠水溶液和饱和食盐水洗涤,分液,硫酸钠干燥,过滤,得到的清液浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到417毫克淡黄色粘稠液体目标化合物。hrms(m+h)forc63h72o12p3:1113.4237(calculated),1113.4256(experimental);(m+na)forc63h71nao12p3:1135.4056(calculated),1135.4063(experimental)。
实施例九:2,2-bis(((3-(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)propanoyl)oxy)methyl)propane-1,3-diylbis(3-(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)propanoate)
以完全类似于实施例八的操作,在氮气保护下,在室温下向含有苯基膦(2.36毫摩尔)的甲苯(20毫升)溶液中加入叔丁醇钠(464毫克)后,再于40分钟内缓缓滴加加入2,4,6-均三甲苯酰氯(431毫克),加毕反应体系继续搅拌2小时后,缓缓加入季戊四醇四丙烯酸酯单体207毫克,此后向反应体系滴加加入0.95毫升的28%浓度双氧水(预先用硫酸调节到ph=2-3),继续搅拌反应1小时后有机相依次用1%碳酸氢钠水溶液和饱和食盐水洗涤,分液,硫酸钠干燥,过滤,得到的清液浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到466毫克淡黄色粘稠液体目标化合物。hrms(m+h)forc81h89o16p4:1441.5101(calculated),1441.5129(experimental);(m+na)forc81h88nao16p4:1463.4921(calculated),1463.4954(experimental)。
实施例十:(((dimethylamino)methyl)(phenyl)phosphoryl)(mesityl)methanone
在氮气保护下,在室温下向含有苯基膦(2.83毫摩尔)的甲苯(30毫升)溶液中加入叔丁醇钠(557毫克)后,再于40分钟内缓缓滴加加入2,4,6-均三甲苯酰氯(520毫克),加毕反应体系继续搅拌2小时后转入一个耐压封管(sealedtube),向封管中加入eschenmosersaltch2=nme2i(524毫克)后继续搅拌1小时,反应液用0.34毫升的28%浓度双氧水在用硫酸调节到ph=2-3条件下处理半小时。有机相依次用1%碳酸氢钠水溶液和饱和食盐水洗涤,分液,硫酸钠干燥,过滤,得到的清液浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到484毫克淡黄色粘稠腊状的目标化合物。hrms(m+h)forc19h25no2p:330.1623(calculated),330.1629(experimental);(m+na)forc19h24nano2p:352.1442(calculated),352.1459(experimental)。
实施例十一:1,3,5-tris(2-hydroxy-3-(phenyl(2,4,6-trimethylbenzoyl)phosphoryl)propyl)-1,3,5-triazinane-2,4,6-trione
在氮气保护下,在室温下向含有苯基膦(2.36毫摩尔)的甲苯(20毫升)溶液中加入叔丁醇钠(464毫克)后,再于40分钟内缓缓滴加加入2,4,6-均三甲苯酰氯(431毫克),加毕反应体系继续搅拌2小时后转入一个耐压封管(sealedtube),向封管中加入233毫克三元环氧(nrc(o))3(这里r是环氧丙烷)继续搅拌过夜,反应液用1.12毫升的28%浓度双氧水在用硫酸调节到ph=2-3条件下处理5小时。有机相依次用1%碳酸氢钠水溶液和饱和食盐水洗涤,分液,硫酸钠干燥,过滤,得到的清液浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯-甲醇梯度洗脱,得到276毫克淡黄色粘稠腊状的目标化合物。hrms(m+h)forc60h67n3o2p3:1114.3938(calculated),1114.3948(experimental);(m+na)forc60h66n3nao12p3:1136.3757(calculated),1136.3762(experimental)。
实施例十二:(3,5-bis(chloromethyl)-2,4,6-trimethylphenyl)(diphenylphosphoryl)methanone
在氮气保护和室温下,将1.5克多聚甲醛固体和5.65克三氯化铝粉末分散在120毫升干燥的二氯乙烷中,体系升温至45度时向其中缓慢滴加3.48克光引发剂tpo在80毫升二氯乙烷中的溶液,维持反应低于回流温度,滴毕半小时后将反应液倒在冰水上,分液,干燥,浓缩,固体用乙酸乙酯重结晶,得到3.43克淡黄色的目标产物,可以直接用于下步反应。
实施例十三:(3,5-bis((bis(2-hydroxyethyl)amino)methyl)-2,4,6-trimethylphenyl)(diphenylphosphoryl)methanone
在氮气保护和室温下,将2.28克实施例十二化合物,1.24克二乙醇胺hn(ch2ch2oh)2,和1.32克三乙胺分散在dmf和乙腈的120毫升(体积比1比6)的混合溶剂中,体系在80度下反应4小时,减压浓缩除去大部分乙腈后,残余物在快速搅拌下倒入100毫升水中,抽滤得到2.66克淡黄色的固体粉末目标产品。1h-nmr(cdcl3):δ=8.00-7.96(m,4h),7.59-7.57(m,2h),7.54-7.50(m,4h),3.73(s,4h),3.50(s,8h),2.60(s,8h),2.52(s,4h),2.04(s,3h),2.00(s,6h);13c-nmr(cdcl3):δ=>220(c=osignal),132.6,131.7,131.6,128.9,128.8,59.7,55.7,53.4,17.8,16.7ppm;hrms(m+h)forc32h44n2o6p:583.2937(calculated),583.2951(experimental);(m+na)forc32h43nan2o6p:605.2756(calculated),605.2774(experimental)。
该化合物的uv-vis吸收光谱图如图2所示(溶剂为乙腈,质量百分浓度),从图可以看出该化合物在uv-led发射波长区域均有显著吸收:
实施例十四:(3,5-bis(hydroxymethyl)-2,4,6-trimethylphenyl)(diphenylphosphoryl)methanone
在氮气保护和室温下,将1.7克实施例十二化合物置于10毫升dmf和10毫升醋酸-醋酸钠缓冲溶液中(ph~8),反应在室温下搅拌过夜后倒在冰水上,分液,干燥,浓缩,固体用乙酸乙酯-己烷在硅胶柱色谱上梯度洗脱得到0.68克淡黄色固体目标产物。hrms(m+h)forc24h26o4p:409.1569(calculated),409.1571(experimental);(m+na)forc24h25nao4p:431.1388(calculated),431.1395(experimental)。
实施例十五:((3-(hydroxymethyl)-2,4,6-trimethylbenzoyl)(phenyl)phosphoryl)(mesityl)methanone
在氮气保护下,将2克多聚甲醛和2.67克三氯化铝溶解在最小量的氯仿中并加热到75度反应45分钟,其后加入8.4克irgacure819光引发剂在氯仿中的饱和溶液,加毕继续反应35分钟,然后补加2.67克三氯化铝,继续反应10分钟(注意用己烷-乙酸乙酯体积比2比1的混合溶剂做展开剂tlc跟踪反应进程),将反应液倒入冰水中,分液,萃取,干燥,过滤,浓缩后残留物在硅胶柱色谱上用己烷-二氯甲烷淋洗剂梯度洗脱,得到2.9克黄色粉末固体目标化合物(同时得到1.7克实施例一化合物)。1h-nmr(cdcl3):δ=7.97-7.91(m,2h),7.59-7.62(m,1h),7.56-7.26(m,2h),6.81(s,2h),6.78(s,2h),5.15(s,2h),2.30(s,1h),2.26(s,3h),2.24(s,3h),2.20(s,6h),2.02(s,6h);13c-nmr(cdcl3):δ=218.8,217.9,169.1,169.0,141.0,139.7,135.7,135.4,135.2,134.9,133.1,131.7,131.6,131.5,129.3,129.2,128.9,128.3,126.8,125.7,73.4,61.3,60.2,21.1,21.0,19.8,19.4ppm;31p-nmr(cdcl3):δ=18.2ppm。hrms(m+h)forc27h30o4p:449.1882(calculated),449.1888(experimental);(m+na)forc27h29nao4p:471.1701(calculated),471.1644(experimental)。
实施例十六:2-(2-hydroxyethoxy)ethyl3-(bis(2,4,6-trimethylbenzoyl)phosphoryl)propanoate
在氮气保护和室温下,根据coenradusjohanneshendrikushendriksen的eth-zürich博士论文中第124页记载的方法合成中间体sodiumbis(2,4,6-trimethylbenzoyl)phosphanide,淡黄色固体粉末。取3.48克该化合物置于100毫升50∶50的thf-水混合溶剂中,依次加入0.062克冰乙酸和1.65克丙烯酸酯单体ch2=chc(o)och2ch2och2ch2oh后反应体系在微波辐照下反应30分钟,不经分离,加入1.6毫升的28%浓度双氧水继续搅拌反应过夜,反应液倒入乙酸乙酯-饱和氯化铵混合溶液中,有机相分离,饱和氯化铵溶液和食盐水分别洗涤一次,干燥,过滤,浓缩,残余物在硅胶柱色谱上用乙酸乙酯-己烷淋洗剂梯度洗脱,得到2.03克黄色固体目标产物。hrms(m+h)forc27h36o7p:503.2199(calculated),503.2207(experimental);(m+na)forc27h35nao7p:525.2018(calculated),525.2034(experimental)。
实施例十七:((hydroxymethyl)phosphoryl)bis(mesitylmethanone)
在氮气保护和室温下,根据coenradusjohanneshendrikushendriksen的eth-zürich博士论文中第124页记载的方法合成中间体sodiumbis(2,4,6-trimethylbenzoyl)phosphanide,淡黄色固体粉末。取3.48克该化合物置于80毫升50∶50的thf-水混合溶剂中,依次加入0.62克冰乙酸和5倍当量的甲醛水溶液后反应体系在微波辐照下反应30分钟,不经分离,加入1.4毫升的28%浓度双氧水继续搅拌反应过夜,反应液倒入200毫升乙酸乙酯-饱和氯化铵混合溶液中,有机相分离,饱和氯化铵溶液和食盐水分别洗涤一次,干燥,过滤,浓缩,残余物在硅胶柱色谱上用乙酸乙酯-己烷淋洗剂梯度洗脱,得到1.88克黄色粘稠蜡状目标产物。hrms(m+h)forc21h26o4p:373.1569(calculated),373.1574(experimental);(m+na)forc21h25nao4p:395.1388(calculated),395.1392(experimental)。
实施例十八:((2-hydroxyethyl)phosphoryl)bis(mesitylmethanone)
在氮气保护和室温下,根据coenradusjohanneshendrikushendriksen的eth-zürich博士论文中第124页记载的方法合成中间体sodiunbis(2,4,6-trimethylbenzoyl)phosphanide,淡黄色固体粉末。取3.48克该化合物置于60毫升thf中,加入0.62克冰乙酸后搅拌反应半小时,离心分离生成的固体,清液转入一个耐压管中,通入3个大气压的环氧乙烷后封闭耐压管,在室温搅拌反应4小时,加入1.4毫升的28%浓度双氧水继续搅拌反应过夜,反应液倒入200毫升乙酸乙酯-饱和氯化铵混合溶液中,有机相分离,饱和氯化铵溶液和食盐水分别洗涤一次,干燥,过滤,浓缩,残余物在硅胶柱色谱上用乙酸乙酯-己烷淋洗剂梯度洗脱,得到1.42克黄色粘稠蜡状目标产物。hrms(m+h)forc22h28o4p:387.1725(calculated),387.1740(experimental);(m+na)forc22h27nao4p:409.1545(calculated),409.1566(experimental)。
实施例十九:bis((bis(2,4,6-trimethylbenzoyl)phosphoryl)methyl)hexane-1,6-diyldicarbamate
在氮气保护下将227毫克的实施例十七化合物和51毫克的己二异氰酸酯hdi置于20毫升的干燥甲苯溶剂中,向体系中加入8毫克的二月桂酸二丁基锡催化剂后在80度加热反应1小时,反应体系浓缩除去甲苯后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到203毫克淡黄色粘稠腊状目标化合物。hrms(m+h)forc50h63n2o10p2:913.3958(calculated),913.3977(experimental);(m+na)forc50h62n2o10nap2:935.3777(calculated),935.3789(experimental)。
实施例二十:bis((bis(2,4,6-trimethylbenzoyl)phosphoryl)methyl)terephthalate
在氮气保护下,将740毫克的实施例十七化合物,202毫克的对苯二甲酰氯,245毫升的三乙胺,和24毫克4-二甲胺基吡啶(dmap)置于60毫升干燥的二氯乙烷溶剂中,室温搅拌反应过夜。混合物用等体积半饱和食盐水洗涤3次,有机相硫酸钠干燥,过滤,浓缩除去溶剂后残余液在硅胶柱色谱上用己烷-乙酸乙酯梯度洗脱,得到604毫克黄色目标化合物。hrms(m+h)forc50h53o10p2:875.3114(calculated),875.3140(experimental);(m+na)forc50h52o10nap2:897.2933(calculated),897.2928(experimental)。
实施例二十一:uv光固化试验
含烯键(丙烯酸酯)样品体系按下列配方制作(以重量百分比计):
双酚a环氧丙烯酸酯(ebecryl605):30%;氨基丙烯酸酯(ebecryl7100):8%;丙氧基化甘油三丙烯酸酯:30%;己二醇二丙烯酸酯:24%;聚硅氧烷丙烯酸酯:0.5%;乙氧基季戊四醇四丙烯酸酯:3.5%;光引发剂:2%本实施例光引发剂,以及2%深圳有为化学公司api-180光引发剂【结构为2-hydroxy-1-(3-(hydroxymethyl)phenyl)-2-methylpropan-1-one】。
对比例体系一:配方与上述相同,除了所用的光引发剂是2%的tpo和2%的irgacure184。
对比例体系二:配方与上述相同,除了所用的光引发剂是4%的irgacure184。
将上述配制实施例和对比例混合物涂覆于卡纸板上形成约30-35微米的涂层,2支80瓦中亚汞灯为光源,变速传送带试验。以指甲反复压刻刮擦不产生印迹为光聚合固化完成的判据。
残余气味测试以5个人分别独立地评价气味级别,评估的标准以数字标示如下:0级:没有气味;1级:非常轻微的气味;2级:轻微的气味;3级:明显的气味;4级:强烈的气味;5级:非常强烈的气味。
结果表明含有实施例化合物零至二,四至十一,以及十三至二十的配方体系均以高于60米/分钟的速度高效固化,且气味等级为0或0-1级;与此对照的,对比例配方体系一的光固化速度为45-50米/分钟,且气味等级为3-4或4;对比例配方体系二的光固化效率在高于30米/分钟的速度下即呈现底干(throughcure)不足问题,且气味等级为4。
上述实验结果表明本发明制备的这些光引发剂具有良好的uv光固化效率性能和低气味特征。
实施例二十二:led光固化试验
含烯键(丙烯酸酯)样品体系按下列配方制作(以重量百分比计):
双酚a环氧丙烯酸酯(ebecryl605):25%;氨基丙烯酸酯(ebecryl7100):8%;丙氧基化甘油三丙烯酸酯:25%;己二醇二丙烯酸酯:22%;聚硅氧烷丙烯酸酯:0.5%;乙氧基季戊四醇四丙烯酸酯:3.5%;光引发剂:2%本实施例光引发剂;助引发剂:14%的cytec公司led-01【巯基修饰的光固化树脂】。
将上述配制实施例混合物涂覆于卡纸板上形成约30-35微米的涂层,1只深圳并日科技公司生产之单位功率为16w/cm2的发射波长为395纳米的led光源(3厘米宽和80厘米长led面光源),变速传送带试验。以指甲反复压刻刮擦不产生印迹为光聚合固化完成的判据。
残余气味测试以5个人分别独立地评价气味级别,评估的标准以数字标示如下:0级:没有气味;1级:非常轻微的气味;2级:轻微的气味;3级:明显的气味;4级:强烈的气味;5级:非常强烈的气味。
结果表明含有实施例化合物一至十一,以及十三至二十的配方体系均以高于45米/分钟的速度高效固化,且气味等级为0或0-1级。
上述实验结果表明本发明制备的这些光引发剂同时具有良好的led光固化效率性能和(净)或低气味特征。
实施例二十三:光固化实验
含烯键(丙烯酸酯)样品体系按下列配方制作(以重量百分比计):
按下述重量百分比(总计100份)配制光固化混合物体系,将此混合物搅拌分散或溶解后喷涂在铝板上(约20-30微米),以一只400瓦高压汞灯为光源辐射引发聚合。实施例使用的是本实施例酰基膦氧化合物光引发剂和深圳有为化学api-1173。固化效率以指压法(即大拇指指甲压膜反复擦拭涂层不破损)判定光固化完成。
白色木器涂料:77.2份pea,5.9份hdda,2.9份tmpta,2.5份实施例一光引发剂,1.5份光引发剂api-1173【结构为2-hydroxy-2-methyl-1-phenylpropan-1-one】,10份tio2。
地板亚光面漆:14.78份高光泽up(vpls2100),14.78份tromoxrkb6,7.39份banc-fixen,1.18份lancowaxhm1666,2.95份tpgda,混合研磨至10微米以下后再加入14.78份高光泽up(vpls2100),5.91份gasilebn,35.47份tpgda,0.06份byk300,1份实施例六光引发剂,1.7份光引发剂api-1173。
光盘油墨:20份pea(eb525),20份eb1710,15份eo-tmpta,10份tpgda,25份tio2,2.5份实施例十五光引发剂,2.5份api-1173光引发剂,3.0份sio2,2.0份消泡剂airex900。
汽车涂料:19.4份聚氨酯丙烯酸酯(沙多玛cn999),19.4份乙氧基双酚a二丙烯酸酯(沙多玛sr601),31份沙多玛sr492,24份沙多玛sr355,0.32份实施例十七光引发剂,2.60份api-180光引发剂,3.28份羟基苯并三唑光吸收剂(注意:本样品的辐照在氮气保护气氛下进行)。
上述含有实施例光引发剂的配方体系均完成了充分固化,展现了优良的光聚合引发活性。
实施例二十四:水性uv-led光固化实验
含烯键(丙烯酸酯)样品体系按下列配方制作(以重量百分比计):
实施例23a:78份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),15份75%tio2颜料,2份助剂丙二醇单甲醚,1份去离子水,2份实施例一光引发剂,2份羟基酮光引发剂api-180。
实施例23b:78份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),15份75%tio2颜料,2份助剂丙二醇单甲醚,1份去离子水,2份实施例六光引发剂,2份羟基酮光引发剂api-180。
实施例23c:96份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),2份实施例一光引发剂,2份羟基酮光引发剂api-180。
实施例23d:96份氰特(现allnex)公司ucecoat7699水性聚氨酯丙烯酸酯分散液(固含量35%wt),2份实施例一光引发剂,2份羟基酮光引发剂api-180。
实施例23e:96份氰特(现allnex)公司ucecoat7177水性聚氨酯丙烯酸酯分散液(固含量35%wt),2份实施例十一光引发剂,2份羟基酮光引发剂api-180。
实施例23f:94份氰特(现allnex)公司ucecoat7856水性聚氨酯丙烯酸酯分散液(固含量35%wt),2份助剂丙二醇单甲醚,2份实施例十三光引发剂,2份羟基酮光引发剂api-180。
实施例23g:96份氰特(现allnex)公司ucecoat7856水性聚氨酯丙烯酸酯分散液(固含量35%wt),2份实施例十五光引发剂,2份羟基酮光引发剂api-180。
实施例23h:96份氰特(现allnex)公司ucecoat7699水性聚氨酯丙烯酸酯分散液(固含量35%wt),1份助剂丙二醇单甲醚,2份实施例十七光引发剂。
实施例23i:96份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),1份助剂丙二醇单甲醚,3份实施例一光引发剂。
实施例23j:96份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),1份助剂丙二醇单甲醚,3份实施例十七光引发剂。
实施例23k:96份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),1份助剂丙二醇单甲醚,3份实施例十三光引发剂。
实施例23l:81份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),12份50%炭黑颜料分散体,2份助剂丙二醇单甲醚,1份去离子水,4份实施例一光引发剂。
实施例23m:83份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),10份水性红色颜料分散体,2份助剂丙二醇单甲醚,1份去离子水,4份实施例十七光引发剂。
对比例23n:78份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),15份75%tio2颜料,2份助剂丙二醇单甲醚,1份去离子水,2份tpo光引发剂,2份光引发剂irgacure184。
对比例23o:94份氰特(现allnex)公司ucecoat7699水性聚氨酯丙烯酸酯分散液(固含量35%wt),2份助剂丙二醇单甲醚,2份光引发剂irgacure500(50∶50的irgacure184和二苯甲酮bp液体混合物)。
对比例23p:96份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),4份irgacure819-dw引发剂。
对比例23q:95份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),1份irgacure184引发剂,4份irgacure819-dw引发剂。
光固化测试方法:将上述配方体系喷涂在基材(玻璃,纸张,塑胶,或木皮)上,在60度加热和抽风条件下除去水分,形成的10-20微米膜体在uv(400w高压汞灯,基材与光源间距为18厘米)或led(16w/cm2单位功率,395纳米波长,基材与光源间距为1厘米)光源下辐照固化。各种基材均置于皮带机上,皮带机线速度设置在30米/分钟。固化硬度用铅笔硬度标示。
储存稳定性考察:配方稳定性(formulation&storagestability)考察通过动态光散射(upa-ex150仪器)和配方在60摄氏度储存10天后粘度上升百分比衡量(与室温下新鲜配制的配方初始粘度相比,ndj-5s数字式粘度计)。配方稳定性的表示方法是:粘度上升百分比(分散体数均平均粒径,单位为纳米;如果形成10微米或以上颗粒,则标记为“沉淀”)。上述配方中水性树脂分散体的室温起始平均粒径典型值在60-100纳米。
残余气味评价:以5个人分别独立地评价气味级别,评估的标准以数字标示如下:0级:没有气味;1级:非常轻微的气味;2级:轻微的气味;3级:明显的气味;4级:强烈的气味;5级:非常强烈的气味。
测试结果如表1。
表1
上述实施例和对比例实验清晰地证明,本发明披露的新型酰基膦氧光引发剂无论是油性还是水性光固化体系,无论是清漆还是含颜料色漆,也无论是uv还是led光源固化,均展现了优秀的和广谱性的光固化实践应用性能,以及突出的环保和健康友好净味等特性,这对绿色印刷,家具木器漆等行业尤为重要;同时,进一步的,由于从技术上欠缺具有足够水溶性或水分散特性的光引发剂,水性uv-led配方体系的复配和储存稳定性是业界长期的困扰性难题,本发明披露的化合物首次揭示了成熟的解决方案,从而有效地解决了这一关键技术挑战。
实施例二十五:水性uv-led喷墨(ink-jet)墨水
水性uv-led喷墨墨水样品按下列配方制作(以重量百分比计):
81份深圳有为化学公司api-313水性聚氨酯丙烯酸酯分散液(固含量43%wt),8份50%炭黑颜料分散体,2份丙二醇单丁醚,2份防沉剂,0.5份byk-021消泡剂,3份去离子水,3.5份实施例一,或实施例五,或实施例六,或实施例十一,或实施例十五,或实施例十六,或实施例十七光引发剂。
将上述配方体系喷涂在铜版纸基材上,在60度加热和抽风条件下除去水分,形成的15微米膜体在uv(400w高压汞灯,基材与光源间距为18厘米)或led(16w/cm2单位功率,395纳米波长,基材与光源间距为1厘米)光源下辐照固化,固化线速度设置在50米/分钟,指甲刮擦法判定表干和里干程度,结果表明上述所有水性黑色喷墨墨水使用无论uv还是节能型led光源均获得了满意的固化效果。
需要强调的是,上述列举的实施例仅是一些示例性试验,不应被视为是限定性试验或条件。本发明申请所涵盖的创新范围应以权利要求书记载为准。
- 还没有人留言评论。精彩留言会获得点赞!