鉴定和分析古DNA样本的方法与流程
本发明涉及生物测序技术领域,具体而言,涉及鉴定和分析古dna样本的方法。
背景技术:
古生物样本对现代生物种群的进化史研究至关重要,古人类基因组的研究成果使人们重新认识到现代人的遗传组成并非只有非洲祖先成分,而是在走出非洲之后又与古尼安德特人和古丹尼索尔人发生过基因交流,颠覆了以往人们对现代人进化史的认识。同时,古生物基因组的研究对现代生物种群,尤其是人类的自然选择和疾病的研究也有着无法替代的重要作用,藏族人的高原适应性基因被证明是来自与古丹尼索尔人的基因组之间的渗透作用。古生物样本作为一种无法复制的遗传资源,对现代生物群体的进化、选择和疾病等研究具有巨大的促进作用而且无法替代。
古生物遗传学研究已经深入到基因组水平。我国作为一个古生物资源大国,不仅有着极其丰富的动植物化石和亚化石资源,更有丰富的古人类样本不断出土,限制我国古人类基因组学发展的最大的瓶颈之一就是缺乏对古dna处理和信息分析方法的总结。
因而,目前鉴定和分析古dna样本的方法仍有待改进。
技术实现要素:
本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明的一个目的在于构建一套基于illumina二代测序数据的古dna的标准信息分析流程,提供一套古人类基因组分析方法。
需要说明的是,本发明是基于发明人的下列发现和工作而完成的:
发明人针对古dna处理和信息分析的方法进行了一系列的理论研究和实验探索,结果发现:
1、古dna的片段化程度很高,因此在构建dna文库的过程中不需对dna进行片段化处理,dna提取完成后可直接进行文库构建。
2、针对古dna,在上机测序时,不宜选择长片段测序,读段长度控制在100bp以内,因为古dna平均长度在50-70bp左右,如果测序时读段的长度超过100bp,一方面会引入大量的接头污染,另一方面会造成大量的数据浪费。
3、针对古dna,原始fastq数据下机后的最重要的一步就是根据illumina数据特征以及古dna的序列特征对数据进行过滤,目的在于最大限度的去除低质量的序列以及被外源污染的dna序列。数据过滤主要包括4方面:对接头进行过滤、对质量值q≤10的低质量碱基进行过滤、对n区(不能识别的区域)进行过滤,以及去除长度小于30bp以及长度大于99bp的读段。如果读段小于30bp,在后续的比对过程中会造成较多的错误比对。因为古dna序列高度片段化,平均长度一般在50-70bp,如果读段过长(大于99bp),则很有可能是来自于现代dna的污染,因此,为了最大限度保留古dna,则应删除这些读读段。这一步及其重要,如果不删除大于99bp的读段,将会影响后续物种鉴定的准确性,这也是跟现代生物样本物种鉴定的一个很大的不同点。
4、为了兼容大部分的比对结果分析,原始下机数据经过质控之后,分别使用soapaligner和bwa对原始古人类dna数据进行比对分析,其中,使用soapaligner比对的数据,最后生成soap格式的比对结果;使用bwa比对的数据,最后生成sam格式的比对结果,并且考虑到古dna的脱氨基作用导致的突变较多,在比对的过程中最多容许4个碱基的错配。由此,比对结果准确可靠,有利于后续分析使用。
5、使用soapsnp和gatk两个软件同时对比对后的数据进行变异检测,主要对单核苷酸变异进行检测;同时,使用soapsnp进行变异检测时,输出cns格式的结果,即把所有位点输出。由此,有利于后续分析使用。
6、针对古dna鉴定:古dna鉴定是进行后续个性化信息分析的最基本前提,发明人综合古dna所具备的分子特征,提出了基于以下2个方面的至少之一进行古dna鉴定的方法:
(1)基于脱氨基突变特征:
古dna的脱氨基突变特征:古生物样本在长期的保存过程中,双链dna会受到一种重要的化学损伤,即胞嘧啶脱氨基。脱氨基作用主要发生在dna片段的端头部位,也就是5’端和3’端。这种脱氨基作用会使胞嘧啶转换成尿嘧啶,因此在文库构建和测序的时候会引入c->t的突变。因此古dna在进行二代测序时,reads的5’端和3’端会出现大量的c->t和g->a的突变。发明人认为,这种突变模式正好可以被利用来鉴定所得序列是否为古dna的证据之一。
(2)基于dna片段化特征:
脱嘌呤作用(dna片段化)特征:脱嘌呤作用是古dna保存过程中发生dna链断裂的一个最重要的化学作用,也就是说,在古dna的片段化过程中,有相当一部分是由于发生了脱嘌呤作用导致的。发明人认为,当将古dna片段比对到参考基因组时,这种脱嘌呤作用就会表现出reads5’末端再往前一个碱基是嘌呤的比例大大增加,相反在3’末端再往后一个碱基是嘧啶的比例会大大增加。因而,发明人认为,古dna这种断裂模式与脱氨基一样,也可以作为鉴定是否为古dna的主要证据之一。
7、发明人还构建了针对女性古dna样本,通过y染色体进行外源dna污染评估的方法:该方法的首先是获得y染色体特定区域(yur,不和其他任何染色体同源而且没有重复序列的区域);然后将所得到的古dna的reads比对到yur,再根据yur和具体的reads数量计算出假设是男性情况下的期望值,最后得到的实际比对上的reads和期望值之间的比值,其即为来自男性的污染率。
由此,在本发明的第一方面,本发明提供了一种获得待测dna样本的dna信息的方法。根据本发明的实施例,该方法包括以下步骤:对所述待测dna样本进行建库和测序,以便获得测序数据,其中,在所述建库时不进行dna片段化的步骤,所述测序读段的长度不超过100bp;对所述测序数据进行过滤处理,以便获得经过过滤处理的测序数据;以及将所述经过过滤处理的测序数据进行比对处理,以便获得比对结果,所述比对结果包含所述待测dna样本的dna信息,其中,所述过滤处理包括下列的至少之一:(1)过滤去除接头序列;(2)过滤去除质量值q≤10的低质量碱基,其中,当所述低质量碱基的数量占整条读段总碱基数量的50%以上时,删除整条读段;当所述低质量碱基在读段的端头,且数量不超过整条读段的50%时,仅切除所述低质量碱基;(3)对n区进行过滤,其中,当读段中含n比例大于10%时,去除所述读段;当n区仅存在于读段两端时,仅切除所述读段两端的n区;(4)去除长度小于30bp及长度大于99bp的读段,所述比对处理最多容许4个碱基的错配。
需要说明的是,本文中所述的“对n区进行过滤,其中,当读段中含n比例大于10%时,去除所述读段;当n区仅存在于读段两端时,仅切除所述读段两端的n区”,其中,“n区”是指不能识别的区域,“含n比例”是指含有不能识别的碱基的比例。
根据本发明的实施例,利用该方法能够有效地基于对待测古dna样本的建库和测序,获得待测古dna样本的dna信息,并且,该信息准确,可信度高,能够有效用于待测古dna的基因组分析,例如变异检测、古dna的鉴定、性别判定以及现代人dna污染率评估。
根据本发明的实施例,同时利用soapaligner和bwa进行所述比对处理。由此,比对结果准确可靠。
根据本发明的一些实施例,利用soapaligner进行所述比对处理时,生成soap格式的比对结果;利用bwa进行所述比对处理时,生成sam格式的比对结果。由此,便于两种比对结果的兼并,最终比对结果可信度高。
在本发明的第二方面,本发明还提供了一种确定待测dna样本是否为古dna的方法。根据本发明的实施例,该方法包括以下步骤:根据前面所述的获得待测dna样本的dna信息的方法,获得待测dna样本的dna信息;基于所述待测dna样本的dna信息,进行变异检测,以便确定所述待测dna样本的变异信息;以及基于所述待测dna样本的变异信息,确定所述待测dna样本是否为古dna,其中,存在下列情形的至少之一是所述待测dna样本为古dna的指示:(1)测序读段呈现如下的脱氨基特征:相对于参考基因组,所述测序读段的5’端和3’端出现大于10%的c->t和g->a的突变;(2)测序读段呈现如下的片段化特征:相对于参考基因组,所述测序读段的5’末端再往前一个碱基是嘌呤的比例显著增加,而3’末端再往后一个碱基是嘧啶的比例显著增加。利用该方法能够有效地进行古dna的鉴定,且结果准确可靠、重复性好。
根据本发明的实施例,同时利用gatk和soapsnp进行所述变异检测。由此,检测结果准确可靠。
根据本发明的一些实施例,利用soapsnp进行所述变异检测时,输出cns格式的结果。由此,便于后续分析。
根据本发明的实施例,所述待测dna样本的变异信息包含单核苷酸变异信息。
在本发明的第三方面,本发明还提供了一种确定古dna样本所属个体的性别的方法。根据本发明的实施例,该方法包括以下步骤:根据前面所述的获得待测dna样本的dna信息的方法,获得待测古dna样本的dna信息;基于所述待测dna样本的dna信息,确定下列性别判定参数的至少之一:比对到x染色体的测序读段和比对到y染色体的测序读段的数量比、比对到x染色体的测序读段和比对到8号染色体的测序读段的数量比,各染色体的测序深度,以及各染色体的杂合子比率;以及基于所述性别判定参数的至少之一,确定所述待测古dna样本所属个体的性别,其中:(1)比对到x染色体的测序读段和比对到y染色体的测序读段的数量比接近9:1,是所述待测古dna样本所属个体为男性的指示;比对到x染色体的测序读段和比对到8号染色体的测序读段的数量比接近1:1,是所述待测古dna样本所属个体为女性的指示;(2)y染色体的测序深度与其他染色体的测序深度接近,是所述待测古dna样本所属个体为男性的指示;y染色体的测序深度显著小于其他染色体的测序深度,是所述待测古dna样本所属个体为女性的指示;(3)x染色体的杂合子比率显著小于其他染色体的杂合子比率,是所述待测古dna样本所属个体为男性的指示;x染色体的杂合子比率并不显著小于其他染色体的杂合子比率,是所述待测古dna样本所属个体为女性的指示。利用该方法能够有效地对古dna样本进行所属个体性别鉴定,并且,结果准确可靠,重复性好。
在本发明的第四方面,本发明还提供了一种确定女性古dna样本中的男性现代dna污染率的方法。根据本发明的实施例,该方法包括以下步骤:
根据前面所述的获得待测dna样本的dna信息的方法,获得待测古dna样本的dna信息;
假设所述女性古dna样本来源于男性,并基于所述待测古dna样本的dna信息,确定测序读段比对到y染色体特定区域的期望比例,其中所述测序读段比对到y染色体特定区域的期望比例的计算公式为:
r=(比对到y染色体特定区域的测序读段数量/比对到基因组的测序读段数量)×0.5;以及
基于所述测序读段比对到y染色体特定区域的期望比例,确定所述待测古dna样本的y染色体污染率,所述待测古dna样本的y染色体污染率即为女性古dna样本中的男性现代dna污染率,
其中,所述待测古dna样本的y染色体污染率的计算公式为:
c=(y/r)×(1/n),
其中,c为y染色体污染率比例,y为比对到y染色体特定区域的测序读段数量,r为所述测序读段比对到y染色体特定区域的期望比例,n为比对到基因组的测序读段总数。
根据本发明的实施例,利用该方法能够有效地确定女性古dna样本中的男性现代dna污染率法。并且,该方法重复性好,结果准确可靠。
根据本发明的实施例,通过下述方法得到所述y染色体特定区域:将人类参考基因组的y染色体基因序列分割成30bp左右长度的人工读段集合;将所述人工读段集合与所述人类参考基因组不包含y染色体的部分进行比对,以便获得经过比对的人工读段;针对所有经过比对的人工读段,仅保留出现3个碱基以上的比对错误的人工读段,然后再去除包含重复序列区域的人工读段,则剩余的所有人工读段组成所述y染色体特定区域。
根据本发明的一些具体示例,所述人类参考基因组为hg19。
此外,还需要说明的是,根据本发明的实施例,本发明的方法具有下列优点的至少之一:
1、本发明的确定古dna样本所属个体的性别的方法,可以提供对古生物样本的性别判定,该性别判定可以扩展到人类和其他所有具有性染色体的动物种类。这里的性别判定不同于现代生物的性别判定,而是基于发明人发现和总结的古dna特征的专有鉴定方法。
2、本发明的确定女性古dna样本中的男性现代dna污染率的方法,能够检测古dna测序数据的现代dna污染率,该方法对于古dna分析的意义重大,因为只有准确评估现代dna污染率,才可进行古dna后续分析。
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
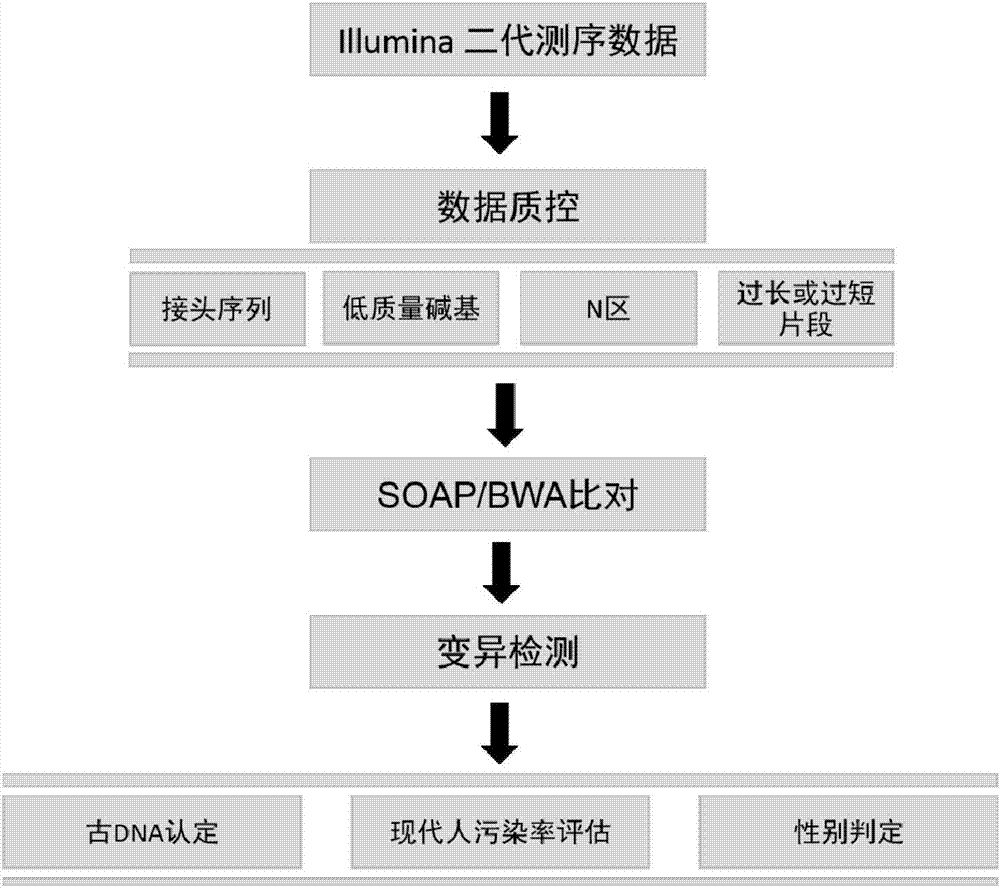
图1是根据本发明实施例的古人类dna信息分析流程图;
图2是实施例1中古人类骨骼样本的dnadamage分析图,
其中,
a图为dna片段化分析结果,灰色框内表示的是古dna片段的碱基,灰色框外表示的是古dna片段5’端最前一个碱基之前的序列和3’端最后一个碱基之后的序列,
b图和c图是脱氨基分析结果,两幅图横坐标表示的是dna片段上的碱基位置,方向为5'-3',b图中的0-25表示dna5'端的前25个碱基,c图的25-0表示dna片段3'端最后的25个碱基;纵坐标表示的是百分比;
图3是实施例1中各染色体中杂合子所占碱基总数的百分比;
图4是实施例1中比对到8号染色体和性染色体的reads(即测序读段)数比较;
图5是实施例1中头发样本测序深度分布情况,纵轴表示深度,横轴表示染色体。
具体实施方式
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
一般方法:
根据本发明的实施例,按照本发明的方法,参照图1,对待测古dna样本进行标准化信息分析,一般包括以下步骤:
1、获得待测dna样本的dna信息
具体步骤如下:
对所述待测dna样本进行建库和测序,以便获得测序数据,其中,在所述建库时不进行dna片段化的步骤,所述测序读段的长度不超过100bp;
对所述测序数据进行过滤处理,以便获得经过过滤处理的测序数据;以及
同时利用soapaligner和bwa,将所述经过过滤处理的测序数据进行比对处理,以便获得比对结果,所述比对结果包含所述待测dna样本的dna信息,
其中,
所述过滤处理包括下列的至少之一:
(1)过滤去除接头序列;
(2)过滤去除质量值q≤10的低质量碱基,其中,当所述低质量碱基的数量占整条读段总碱基数量的50%以上时,删除整条读段;当所述低质量碱基在读段的端头,且数量不超过整条读段的50%时,仅切除所述低质量碱基;
(3)对n区进行过滤,其中,当读段中含n比例大于10%时,去除所述读段;当n区仅存在于读段两端时,仅切除所述读段两端的n区;
(4)去除长度小于30bp及长度大于99bp的读段,
所述比对处理最多容许4个碱基的错配,
利用soapaligner进行所述比对处理时,生成soap格式的比对结果;利用bwa进行所述比对处理时,生成sam格式的比对结果。
2、确定待测dna样本是否为古dna的方法
具体步骤如下:
基于所述待测dna样本的dna信息,同时利用gatk和soapsnp进行变异检测,以便确定所述待测dna样本的变异信息;以及
基于所述待测dna样本的变异信息,确定所述待测dna样本是否为古dna,
其中,存在下列情形的至少之一是所述待测dna样本为古dna的指示:
(1)测序读段呈现如下的脱氨基特征:相对于参考基因组,所述测序读段的5’端和3’端出现大于10%的c->t和g->a的突变;
(2)测序读段呈现如下的片段化特征:相对于参考基因组,所述测序读段的5’末端再往前一个碱基是嘌呤的比例显著增加,而3’末端再往后一个碱基是嘧啶的比例显著增加,
利用soapsnp进行所述变异检测时,输出cns格式的结果,
所述待测dna样本的变异信息包含单核苷酸变异信息。
3、确定古dna样本所属个体的性别
具体步骤如下:
基于所述待测dna样本的dna信息,确定下列性别判定参数的至少之一:比对到x染色体的测序读段和比对到y染色体的测序读段的数量比、比对到x染色体的测序读段和比对到8号染色体的测序读段的数量比,各染色体的测序深度,以及各染色体的杂合子比率;以及
基于所述性别判定参数的至少之一,确定所述待测古dna样本所属个体的性别,其中:
(1)比对到x染色体的测序读段和比对到y染色体的测序读段的数量比接近9:1,是所述待测古dna样本所属个体为男性的指示;比对到x染色体的测序读段和比对到8号染色体的测序读段的数量比接近1:1,是所述待测古dna样本所属个体为女性的指示;
(2)y染色体的测序深度与其他染色体的测序深度接近,是所述待测古dna样本所属个体为男性的指示;y染色体的测序深度显著小于其他染色体的测序深度,是所述待测古dna样本所属个体为女性的指示;
(3)x染色体的杂合子比率显著小于其他染色体的杂合子比率,是所述待测古dna样本所属个体为男性的指示;x染色体的杂合子比率并不显著小于其他染色体的杂合子比率,是所述待测古dna样本所属个体为女性的指示。
4、确定女性古dna样本中的男性现代dna污染率
具体步骤如下:
假设所述女性古dna样本来源于男性,并基于所述待测古dna样本的dna信息,确定测序读段比对到y染色体特定区域的期望比例,其中所述测序读段比对到y染色体特定区域的期望比例的计算公式为:
r=(比对到y染色体特定区域的测序读段数量/比对到基因组的测序读段数量)×0.5;以及
基于所述测序读段比对到y染色体特定区域的期望比例,确定所述待测古dna样本的y染色体污染率,所述待测古dna样本的y染色体污染率即为女性古dna样本中的男性现代dna污染率,
其中,所述待测古dna样本的y染色体污染率的计算公式为:
c=(y/r)×(1/n),
其中,c为y染色体污染率比例,y为比对到y染色体特定区域的测序读段数量,r为所述测序读段比对到y染色体特定区域的期望比例,n为比对到基因组的测序读段总数。
其中,通过下述方法得到所述y染色体特定区域:将人类参考基因组的y染色体基因序列分割成30bp左右长度的人工读段集合;将所述人工读段集合与所述人类参考基因组不包含y染色体的部分进行比对,以便获得经过比对的人工读段;针对所有经过比对的人工读段,仅保留出现3个碱基以上的比对错误的人工读段,然后再去除包含重复序列区域的人工读段,则剩余的所有人工读段组成所述y染色体特定区域。所述人类参考基因组为hg19。
实施例1
根据上述“一般方法”所示的本发明的方法,对待测古dna样本进行标准化信息分析,具体如下:
其中,待测古dna样本2例:1例古人类骨骼样本和1例古人类毛发样本。该2例古生物样本均由中国科学院古脊椎动物与古人类研究所提供,出土年代大约在3000-8000年前,其中1例为古人类骨骼样本(human_bone),1例为古人类毛发样本(human_hair)(见表1)。
具体过程如下:
一、illumina二代测序数据的获取
本发明基于illumina二代测序数据,2例古生物样本的dna提取和建库方法详见:
[1]n.rohland,m.hofreiter.ancientdnaextractionfrombonesandteeth[j].natureprotocols,2007,2(7):1756-1762.doi:10.1038/nprot.2007.247;
[2]m.t.gansauge,m.meyer.single-strandeddnalibrarypreparationforthesequencingofancientordamageddna[j].natureprotocols,2013,8(3):737-748.doi:10.1038/nprot.2013.038,
通过参照将其全文并入本文。
古人类样本所采用的测序策略采用illuminahiseq2000pe50,每个样本的原始下机fastq格式的测序数据量详见表1。其中,在建库过程中,毛发样本对脱氨基作用产生的尿嘧啶进行了去除,骨骼样本未对尿嘧啶做处理。骨骼和毛发样本最后测序数据量均为15gb。
二、原始下机fastq数据的质控
本发明严格按照技术方案中描述的数据过滤方法,对2例古人类骨骼和毛发样本的原始下机fasta数据进行了严格的过滤。具体执行标准如下:1)如果发现读段中包含有接头序列,切除接头序列部分;2)如果质量值q≤10的碱基数占整条读段总碱基数量的50%以上时,删除整条读段,如果低质量碱基在读段的端头,且数量不超过整条读段的50%,则仅切除低质量部分的碱基;3)去除含n比例大于10%的读段,如果n区仅存在于读段两端,仅切除读段两端的n区,其余碱基保留;4)去除长度小于30bp以及长度大于49bp的读段。过滤后的数据量详见表2。
三、比对分析
原始下机数据经过质控之后,分别使用soapaligner和bwa对原始古人类骨骼样本和毛发样本dna数据进行比对分析,参考基因组使用的版本为人类hg19。
soapaligner比对的命令参数如下:
毛发样本:
soap–dhg19.fa.index–ahuman_hair.fq1.gz–bhuman_hair.fq2.gz-ohuman_hair.soap-2human_hair.single–uhuman_hair.unmapped-n5-r1-l30-s30-v2-p4-m0-x80
骨骼样本:
soap–dhg19.fa.index–ahuman_hair.fq1.gz–bhuman_hair.fq2.gz-ohuman_hair.soap-2human_hair.single–uhuman_hair.unmapped-n5-r1-l30-s30-v4-p4-m0-x80
bwa比对的命令参数如下:
毛发样本:
bwaalnhg19.fahuman_hair.fq1.gz-l30-k2-t4-q15-i>human_hair.fq1.sai;bwaalnhg19.fahuman_hair.fq2.gz-l30-k2-t4-q15-i>human_hair.fq2.sai;bwasampe-a80hg19.fahuman_hair.fq1.saihuman_hair.fq2.saihuman_hair.fq1.gzhuman_hair.fq2.gz>human_hair.sam
骨骼样本:
bwaalnhg19.fahuman_hair.fq1.gz-l30-k4-t4-q15-i>human_hair.fq1.sai;bwaalnhg19.fahuman_hair.fq2.gz-l30-k4-t4-q15-i>human_hair.fq2.sai;bwasampe-a80hg19.fahuman_hair.fq1.saihuman_hair.fq2.saihuman_hair.fq1.gzhuman_hair.fq2.gz>human_hair.sam
比对完成后,提取比对结果中的unique比对的reads,同时过滤掉低质量的比对结果和没有配对的比对结果,用于下一步分析。结果的数据信息详见表2和表3。其中中国古人类骨骼样本的测序结果只有极少量数据比对到了人类基因组(0.1%~0.2%),这些数据不足以支持变异检测等后续信息分析。因此对骨骼样本的信息分析仅限于过滤、比对和dnadamage分析。中国古人类头发样本过滤后的比对率达到了10%,数据足够支持后续snpcalling等信息分析,因此发明人对此样本的测序结果进行了较为全面的信息分析,包括过滤、比对、dnadamage分析、深度和覆盖度分析、snpcalling分析、性别判定分析和现代人污染率分析等。
四、变异检测
本发明同时使用gatk和soapsnp对古人类毛发样本进行变异检测。
gatk:
使用gatk进行变异检测完全按照gatk的操作流程进行,具体可参照https://www.broadinstitute.org/gatk/。使用gatk进行变异检测首先对bwa比对生成的sam文件按照染色体组型(karyotypic)进行重新排序;然后将sam格式的比对文件转换成bam格式;将bam文件中的条目按照物理位置从小到大进行排序;对重复出现且比对到染色体同一位置的reads进行标记;对比对到indel区域的read进行重比对;对碱基质量值进行校正,最后生成human_hair.bam和human_hair.metrics;最后使用unifiedgenotyper进行变异检测。具体的参数如下:
java–jargenomeanalysistk.jar-glmsnp-linfo-rhg19.fa-tunifiedgenotyper-ihuman_hair.bam-ddbsnp_137.hg19.vcf-ohuman_hair.vcf-metricshuman_hair.metrics-stand_call_conf10
-stand_emit_conf30。
soapsnp:
使用soapsnp进行变异检测首先也是对soapaligner的比对结果按照染色体组型(karyotypic)进行重新排序,然后在同一条染色体内按照物理位置从小到大进行排序。具体参数如下:
soapsnp–ihuman_hair.soap.gz–dhg19.fa–ohuman_hair.cns-r0.0001-t-u-l49-m-mhuman_hair.mat
五、古dna认定
由于在对古人类毛发样本进行单链dna文库构建的过程中,发明人使用了一种特殊的酶udg将尿嘧啶移除了,以防止c->t的突变对后续分析造成结果的不准确。这样在单链方法构建的文库的结果无法看出很明显的dnadamage模式。古人类骨骼样本在建库过程中并未使用udg做处理,因此,在做古dna认定时,发明人使用古人类骨骼样本。
发明人使用mapdamage对古人类骨骼样本的测序结果的错配模式和片段化模式进行了统计分析和绘图,具体使用参数入下:
perlmapdamage-0.3.3.plmap–ihuman_bone.sam–ddirectory–rhg19.fa-c-thair-l49;perlmapdamage-0.3.3.plmerge–ddirectory;mapdamage-0.3.3.plplot–ddirectory
结果如图2所示,从片段化模式来看,5’端嘌呤的比例显著增加,而嘧啶的比例则相应显著降低;从脱氨基模式看,5’端积累了大量的c->t突变,而3’端则相应积累了大量的g->a的突变。因此,无论是片段化模式还是脱氨基特征都完全符合古dna特征,因此发明人可以确定发明人得到的测序数据为古dna。
六、性别判定
发明人从3个方面对头发样本所属古人个体进行了性别判定分析:
1:分析假设:如果头发样本所属古人是一个男性个体,那么x染色体上的杂合子比例要远远小于其染色体。
分析结果:x染色体中杂合子比率并没有显著小于其他染色体(见图3)。结果为女性。
2:分析假设:x染色体和y染色体有效长度比例为9:1,x染色体和8号染色体比例接近1:1。如果是男性,那么比对到x染色体的reads数量和y染色体的reads数量应该接近9:1;如果是女性,x染色体与8号染色体之间的比例应该接近1:1。
分析结果:mapping到x染色体与8号染色体的reads数比例接近1:1,而x染色体与y染色体的比例为40:1,远远大于9:1(见图4)。结果为女性。
3:分析假设:如果是男性,y染色体的测序深度应该与其他染色体接近。
分析结果:y染色体测序深度明显小于其他染色体各个区域(见图5)。结果为女性。
综合以上三方面的结果,毛发样本所属古人个体为一个女性个体。
七、现代人dna污染率评估
由于发明人测序的样本只有1个个体,而且发明人无法得知该古人与其他古人类以及现代人之间的亲缘关系,测序数据量又较少。因此无法找到该古人特有的segregating位点,无法使用mtdna和常染色体数据进行现代人污染率的评估。但由于发明人判断该个体为女性个体,因此可以进行现代男性个体的污染率评估。基本原理是将所得到的reads比对到y染色体特有区域(yur,不和其他任何染色体同源而且没有重复序列的区域),再根据yur和具体的reads数量计算出假设是男性情况下的期望值,最后实际比对上的reads和期望值之间的比值就是来自男性的污染率。
最后得到的现代男性污染率为1.72%~5.98%,与其他古人类dna文献报道污染率相比偏高,由于所得到的实际数据量偏低,可能会造成一定程度的低估或者高估。在后续分析中需要对可能来自现代人的reads进行充分过滤以保证结果的可靠性。
表12例古生物测序数据情况
表2古人类头发样本的过滤和比对分析结果
表3古人类骨骼样本的过滤和比对分析结果
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不一定指的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。
尽管已经示出和描述了本发明的实施例,本领域的普通技术人员可以理解:在不脱离本发明的原理和宗旨的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由权利要求及其等同物限定。
- 还没有人留言评论。精彩留言会获得点赞!