一类新型的锰催化乙醇缩合制备丁醇的方法与流程
本发明涉及一类新型的锰催化乙醇缩合制备丁醇的方法,属于催化工艺技术领域。
背景技术:
能源是人类生存和文明发展的重要物质基础,在为我们提供能量和动力的同时,也是众多基础化学品的最重要来源。目前生产生活所需要的化学品和燃料主要利用化石资源转化而来,这种非可再生资源的日渐消耗促使人们发展其它替代性的可再生碳源进行燃料及化学品的生产。而通过生物转化而获得的生物燃料是可再生能源开发利用的重要方向,具有良好的可贮藏性和可运输性,可提供替代石油的液体燃料。2015年全球共生产了高达相当于7490万吨原油能量的生物质燃料,其中超过70%的生物燃料是通过生物质发酵工艺而获取的生物乙醇。
目前生物乙醇的主要是用途是与汽油混合直接用于未经改造的内燃机中,从而有效降低石油资源的消耗。乙醇作为生物燃料有许多缺点,首先由于乙醇水溶性容易吸湿,汽油必须在临近使用前进行乙醇和汽油的混合,从而造成储存和运输过程复杂的问题。其次,乙醇在燃烧过程中会产生乙酸,对金属部件特别是铜具有腐蚀性,另外乙醇的能量密度较低,当使用高比例的含乙醇汽油会造成燃烧值和动力性下降的问题,以上这些缺点都会限制乙醇作为混合添加剂应用在汽油中。而使用高级醇,例如,丁醇和更加长链的醇,由于丁醇等长链醇具有更高的能量密度并具有较低的蒸气压,能以更高比例与汽油混合甚至单独作为汽油内燃机的燃料使用。尽管丁醇等长链醇是更为优质的燃料,然而目前尚缺乏高效的方法通过生物质资源直接合成长链醇。已知最好的生物合成的方法,即所谓的abe发酵法,仅会以低的产率得到丙酮,丁醇和乙醇的混合物,由于分离困难,且成本较高,因此被以丙烯为原料的化学合成方法所取代。
基于全球范围内生物质乙醇的大规模生产,发展一种有效的将乙醇直接转化为长链醇的生产工艺将成为由生物质资源制取长链醇类优质生物燃料的重要技术。从某种意义上说,guerbet反应是一种由乙醇合成丁醇或者长链醇的非常理想的方法,因为对于该类反应来说其副产物仅仅是水。而利用乙醇作为guerbet反应的底物是非常具有挑战性的。首先,乙醇的脱氢反应是热力学十分不利的反应过程,δg0=12.87kcal/mol;其次,反应中间体乙醛的aldol缩合反应也时常面临选择性差的问题。目前,只有少数的一些例子报道了guerbet反应,其中非均相催化剂通常需要较为苛刻的反应条件或者只得到低的丁醇选择性。与其相比,尽管使用均相催化剂催化该反应通常下可以得到更高的活性,更好的选择性。但到目前为止有关均相催化guerbet反应的报道都主要集中在ru和rh等贵金属上(图1)。
利用廉价金属催化剂替代贵金属催化剂是近年来催化科学研究领域的一个重要发展方向。然而,到目前为止均相廉价金属催化的乙醇等低碳醇类小分子的碳-碳偶联反应制备长链醇类化合物还未见报道。因此,发展相应的高效均相廉价金属催化剂实现(生物)乙醇等低碳醇类小分子的碳-碳偶联反应将成为制备优质生物燃料的一个关键技术。
技术实现要素:
本发明提供了一类新型的锰催化乙醇偶联制备丁醇的反应,所采用的锰络合物是一种基于廉价金属锰的nnp或者pnp络合物,该反应表现出了很高的催化活性(ton=114120,其中前12h的tof为3078h-1)和92%的丁醇选择性,因此,本发明提供的新方法将对生物质资源高效利用,特别是对优质生物燃料生产、生物质平台化合物的催化活化和转化及相应廉价、高效催化剂的设计,提供重要的技术支持和理论指导。
本发明首先提供的锰络合物,其结构式如式i、式ii、式iii或式iv所示:
式i、式ii和式iii中,
其中,x2为n或p,r为甲基、乙基、异丙基、叔丁基、环戊基、环己基、苯基或2-吡啶基;r1为h、甲基、乙基、苄基或2-吡啶甲基;
式i中,x和x1均独立地选自h、cl、br、i、co(羰基)、oac(醋酸根)、oet(乙氧基)、ome(甲氧基)、obn(苄氧基)、oh和otf(三氟甲磺酰基),且x和x1不同时为co。
所述nnp配体的结构式如式v或式vi所示:
式v和式vi中,a表示c或n;
式v中,r表示为甲基、乙基、异丙基、叔丁基、环戊基、环己基或苯基;
r1表示为h、甲基、乙基、苄基或2-吡啶甲基;
r3、r4、r5和r6均独立地表示h、c1~c6烷基、c1~c6烷氧基、苯基、c1~c6烷基取代苯基、卤素、苄基、羟基、萘基、呋喃基或噻吩基;
式vi中,r7、r8和r9均独立地表示h、c1~c6烷基、c1~c6烷氧基、苯基、c1~c6烷基取代苯基、卤素、苄基、羟基、萘基、呋喃基或噻吩基。
所述pnp配体的结构式如式vii所示:
式vii中,r表示为甲基、乙基、异丙基、叔丁基、环戊基、环己基或苯基;
r1表示为h、甲基、乙基、苄基或2-吡啶甲基。
本发明中,c1~c6烷基具体指的是甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、仲戊基、叔戊基、环戊基、正己基、异己基、新己基、仲己基、叔己基或环己基。
c1~c6烷氧基为甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基、正戊氧基、异戊氧基、新戊氧基、仲戊氧基、叔戊氧基、环戊氧基、正己氧基、异己氧基、新己氧基、仲己氧基、叔己氧基或环己氧基。
本发明所述锰络合物具体如式1-式16所示,优选式2、式3、式8、式11和式12:
各式中,tbu表示叔丁基,ph表示苯基,cy表示环己基,ipr表示异丙基。
本发明进一步提供了所述锰络合物的制备方法,包括如下步骤:
在惰性气氛的保护下,五羰基卤化锰盐与所述nnp配体或所述pnp配体进行反应,即得所述锰络合物;
所述五羰基卤化锰盐的化学式为mn(co)5x3,其中,x3表示cl、br或i。
上述的制备方法中,所述五羰基卤化锰盐与所述nnp配体或所述pnp配体的摩尔比可为1:1.05~1.1,如1:1.1;
所述反应的温度可为70~110℃,时间可为6~20小时,如在70℃下反应8~20小时、8小时、15小时或20小时。
上述的制备方法中,所述反应在溶剂中进行,所述溶剂可为甲苯、二甲苯和四氢呋喃中至少一种。
本发明锰络合物能够催化乙醇的guerbet反应,获得的丁醇及其更高级醇的化合物可以作为汽油中的添加剂。
所述guerbet反应的反应方程式具体如式(1)
其中,[mn]表示所述锰络合物;
所述guerbet反应中,所述锰络合物的摩尔用量可为乙醇的0.00001%~0.01%,具体可为0.01%,所述乙醇的的摩尔用量可50mmol~1300mmol;
所述guerbet反应的温度可为100~180℃,时间可为2~168小时,如在160℃下反应24小时;
所述guerbet反应在乙醇钠存在的条件下进行;
所述guerbet反应的体系中所述乙醇钠的浓度可为0.1~2m,具体可为1m。
基于本发明所述锰络合物,提供了一种基于廉价金属锰催化的乙醇偶联制备丁醇的方法,表现出了很高的催化活性(ton=114120,其中前12h的tof为3078h-1)和92%的丁醇选择性(图1);本发明将对生物质资源高效利用,特别是对优质生物燃料生产、生物质平台化合物的催化活化和转化及相应廉价、高效催化剂的设计,提供重要的技术支持和理论指导。
本发明取得了如下技术效果:
1、本发明提供报道一类新的锰-nnp或者pnp络合物催化乙醇缩合成丁醇的反应,能够高选择性的合成丁醇化合物,所述络合物具有高催化活性,具有很高的研究价值和应用前景。
2、本发明还提供了一种制备锰-nnp或者pnp络合物的方法,通过三齿的nnp或者pnp配体与锰盐在溶剂中加热搅拌反应,过滤所得沉淀即是新型锰-nnp或者pnp络合物,制备简单,易操作。
附图说明
图1为可实现乙醇的guerbet反应的均相催化剂及其催化效果。
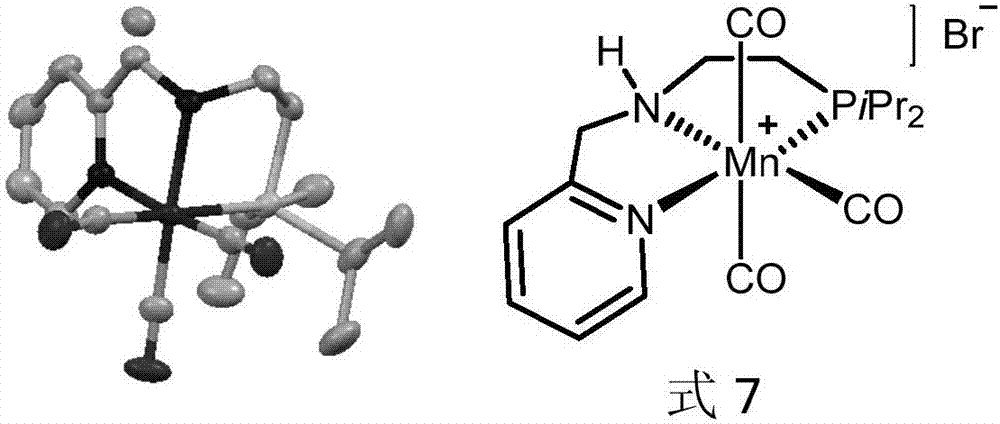
图2为式7所示锰络合物的结构式及其x-射线单晶结构。
图3为式9所示锰络合物的结构式及其x-射线单晶结构。
图4为式11所示锰络合物的结构式及其x-射线单晶结构。
图5为式15所示锰络合物的结构式及其x-射线单晶结构。
具体实施方式
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
下列实施实例中,me表示甲基,tbu表示叔丁基,nbu表示正丁基,ph表示苯基,bn表示苯甲基,cy表示环己基,ipr表示异丙基,pe表示石油醚,ea表示乙酸乙酯,tlc表示薄层色谱,nmr表示核磁共振。
下述实施例中的配体双[2-(二叔丁基膦基)乙基]胺(tbupnp)、双[2-(二环己基膦基)乙基]胺(cypnp)和双[2-(二异丙基膦基)乙基]胺(iprpnp),从alfa-aesar试剂公司购买,纯度>95%;其余配体均参考文献合成:(organometallics2003,22,445.;organometallics,2009,28,6331.;organometa-llics2012,31,5239.;daltontrans.,2011,40,10397.)。
mn(co)5br从alfa-aesar试剂公司购买,纯度>97%,直接使用。
所用溶剂均从上海国药试剂公司购买,使用前用标准方法提纯干燥。
一、py-nnp配体的制备:
1、合成2-氯代-n,n-二(三甲基硅基)乙基胺
往史莱克瓶中先后加入2-氯代乙胺盐酸盐(4.6g,40mmol),net3(18ml,132mmol)和的二氯甲烷50ml。反应在室温搅拌的情况下,往该体系加入三甲基氯硅烷(90mmol,9.8g,11.4ml)的二氯甲烷溶液(20ml)。反应室温搅拌过夜,反应结束后,减压除去过量的三乙胺,三甲基氯硅烷和二氯甲烷,往所得的剩余物中加入60ml的正己烷,反应室温搅拌30分钟,过滤除去net3·hcl。滤液浓缩后,经过减压蒸馏得到目标产物(6.0g,68%)。1hnmr(400mhz,cdcl3)δ3.37–3.21(m,2h),3.18–2.99(m,2h),0.14(s,18h).13cnmr(101mhz,cdcl3)δ47.38,44.83,1.89.
2、合成2-异丙基膦乙胺硼烷络合物
在氩气条件下,往100ml的史莱克瓶中,加入二异丙基膦(2.92g,20mmol)和正己烷(20ml),将上述混合液冷却到-78度,往该体系中逐滴加入正丁基锂(2.5m,8.8ml,22mmol)。加完后,反应升至室温,继续搅拌1h后,得到lipipr2白色悬浊物。
在氩气保护下,将2-氯代-n,n-二(三甲基硅基)乙基胺(4.46g,20mmol)和20ml正己烷,加入到史莱克瓶中,在搅拌情况下,往该混合物加入上述lipipr2的悬浊液。反应室温搅拌30min后,加热回流8h。反应结束后,往反应中加入10ml脱气水和2m10ml的h2so4溶液,反应室温搅拌一小时,之后往该反应中加入11ml4mnaoh溶液,反应继续搅拌一小时。分离有机相,水相用正己烷萃取(2×15ml),合并有机相,无水硫酸钠干燥,过滤,抽干后得粗品2-异丙基膦乙胺。
将所得到的2-异丙基膦乙胺溶解于脱气的thf(30ml)中,往该溶液中加入1m的硼烷四氢呋喃溶液,反应室温搅拌2小时后,抽干,剩余物经柱层析分离得到目标产物(0.75g,41%)。1hnmr(400mhz,cdcl3)δ2.98(td,j=8.7,6.2hz,2h),1.99(ddt,j=14.3,10.4,7.1hz,1h),1.84–1.69(m,2h),1.35(s,2h),1.18(ddd,j=10.9,7.1,4.3hz,12h),0.82–0.11(brs,3h).δ13cnmr(101mhz,cdcl3)δ37.55,23.46(d,j=29.1hz),22.08(d,j=33.5hz),16.91(d,j=20.3hz).31pnmr(162mhz,cdcl3)δ29.42(q).hr-ms(esi)calcd.forc14h29bn2p[m+h]+:176.1734;found:176.1720.
3-1、合成py-nnp配体的硼烷络合物
在氩气条件下,往史莱克瓶中,加入2-吡啶甲醛(0.44g,4.14mmol)和5mlthf,往该溶液中加入2-异丙基乙胺硼烷络合物(0.73g,4.14mmol)的thf(5ml)溶液。反应搅拌0.5h,将thf溶剂抽干,所得吡啶-imine-nnp配体的硼烷络合物不经过纯化直接投入下一步反应中。
在冰水浴条件下,往上述所得的吡啶-imine-nnp配体的硼烷络合物中加入20ml甲苯,往该溶液中缓慢滴加1.2m的dibal的甲苯溶液(1.3ml,1.56mmol)。反应室温搅拌0.5小时后,加入3ml水淬灭,甲苯萃取两次(2×5ml),合并有机相。硫酸钠干燥,过滤,将溶剂抽干后,经柱层析分离得到纯的吡啶nnp配体的硼烷络合物(0.65g,59%)。1hnmr(400mhz,cdcl3)δ8.55(d,j=4.8hz,1h),7.64(td,j=7.7,1.8hz,1h),7.37–7.26(m,1h),7.16(dd,j=7.4,4.9hz,1h),3.93(d,j=4.9hz,2h),2.93(dt,j=12.0,7.1hz,2h),1.99(ddt,j=14.3,10.4,7.1hz,3h),1.83(ddd,j=11.0,9.6,8.1hz,2h),1.17(ddd,j=12.1,7.1,5.5hz,12h),0.75–0.14(brs,3h).13cnmr(101mhz,cdcl3)δ159.09,149.31,136.51,122.28,122.05,54.75,44.34,22.11(d,j=33.4hz),19.87(d,j=30.2hz),16.94(d,j=21.8hz).31pnmr(162mhz,cdcl3)δ32.25(q).hr-ms(esi)calcd.forc14h29bn2p[m+h]+:267.2155;found:267.2136.
3-2、合成py-nnp配体
将含有py-nnp配体的硼烷络合物(0.32g,1.2mmol)溶解于脱气的et2nh,将上述溶液在氩气保护下加入20mlschlenk瓶中,反应在95度进行24小时。反应结束后,抽干et2nh,剩余物再氩气保护下过柱分离(淋洗剂比例:石油醚/乙酸乙酯=3:1to石油醚/乙酸乙酯/甲醇=3:1:0.2),得到目标产物(0.22g,72%)。
1hnmr(400mhz,cdcl3)δ8.48(d,j=4.2hz,1h),7.56(td,j=7.7,1.8hz,1h),7.24(d,j=7.8hz,1h),7.08(dd,j=6.7,5.2hz,1h),3.86(s,2h),2.81–2.56(m,2h),2.10(s,2h),1.79–1.60(m,2h),1.60–1.48(m,2h),0.95-1.03(m,12h).13cnmr(101mhz,cdcl3)δ159.09,149.31,136.51,122.28,122.05,54.75,44.34,22.11(d,j=33.4hz),19.87(d,j=30.2hz),16.94(d,j=21.8hz).31pnmr(162mhz,cdcl3)δ-0.91(s).hr-ms(esi)calledforc14h26n2p[m+h]+:253.1828;found:253.1828.
二、n-me-pnp(cy)配体的制备
在氩气保护下,往50ml的schlenk瓶中,加入二环己基膦(1.00g,5mmol)和脱气thf(10ml),将上述溶液冷却至-20度,往上述溶液中缓慢加入nbuli(2.2ml,2.5minhexanes,5.5mmol),加完后缓慢升至室温并回流1小时。在氩气保护下,往另一个50ml的schlenk瓶中加入men(ch2ch2cl)2·hcl(0.49g,2.52mmol)和thf(10ml),将上述溶液冷却至-20度,往上述溶液中缓慢加入nbuli(1.1ml,2.5minhexanes,2.52mmol),加完后缓慢升至室温并继续搅拌2小时。之后将上述溶液在-78度下,缓慢加入的二环己基膦锂的thf溶液,之后反应液缓慢升至室温,并加热回流过夜。反应结束后,抽干thf,往剩余液中加入5ml脱气水,溶液用脱气乙醚萃取三次(3×10ml),合并有机相,无水硫酸钠干燥,过滤,并将有机相浓缩至5ml,放-20度冰箱30min后有大量白色固体析出,低温氩气过滤,得白色固体(0.45g,38%)。1hnmr(400mhz,tol-d8)δ2.71(dt,j=9.4,6.2hz,4h),2.31(s,3h),2.13(dt,j=4.3,2.1hz,4h),1.91–1.74(m,24h),1.56(m,4h),1.25(m,18h).13cnmr(101mhz,tol-d8)δ56.76(d,j=29.4hz),41.65,33.58(d,jc-p=14.9hz),30.51(d,jc-p=15.4hz),29.14(d,jc-p=8.7hz),27.32(d,jc-p=5.5hz),26.63.20.61,20.41,20.22,20.03,19.84,19.76,19.65,19.46.31pnmr(162mhz,tol-d8)δ-8.00(s).hr-ms(esi)calledforc29h56np2[m+h]+:480.3883;found:480.3880.
实施例1、合成吡啶nnp-mn(co)2br络合物(式7所示)
反应方程式如下所示:
在氩气氛围下,往25ml的schlenk瓶中,加入[mn(co)5br](129mg,0.47mmol),[py-nnp-ipr]l5(132mg,0.52mmol)和脱气thf(10ml),反应室温搅拌过夜。随着反应时间的进行,溶液逐渐由淡黄色固体析出。反应结束后,氩气过滤,用脱气乙醚淋洗滤饼,真空干燥后得浅黄色固体(192mg,87%)。
核磁:1hnmr(400mhz,cd2cl2)δ8.62(d,j=4.9hz,1h),8.20(s,1h),7.72(d,j=7.0hz,1h),7.33(d,j=6.8hz,1h),7.29–7.25(m,1h),4.79(d,j=18.7hz,1h),3.88(d,j=17.8hz,1h),3.11–2.79(m,2h),2.41(m,1h),2.08(t,j=13.6hz,1h),1.62–0.94(m,14h).13cnmr(101mhz,cd2cl2)δ162.54,152.49,138.92,124.66,121.87,59.99,29.67,24.43(d,j=17.0hz),24.05,23.84(d,j=17.8hz),20.26,19.32(d,j=2.3hz),18.85,17.82(d,j=6.0hz).31pnmr(162mhz,cd2cl2)δ77.54(s).
高分辨:hr-ms(esi)calledforc17h25mnn2o3p[m]+:391.0978;found:391.0979.
红外:ir:3029,2905,2018,1924,1459,1104,1056,779,682,640,618cm-1.
x-射线单晶结构如图2所示。
由上述分析结果可知所制备的目标锰络合物结构正确。
实施例2、合成n-me-pnp(ipr)-mn(co)2br络合物(式9所示)
在氩气氛围下,往25ml的schlenk瓶中,加入[mn(co)5br](120mg,0.44mmol),[n-me-pnp-ipr](153mg,0.48mmol)和脱气toluene(10ml),反应加热至100度,搅拌过夜。反应结束后,抽干甲苯,加入10ml脱气正己烷,氩气过滤,滤饼真空干燥后得浅黄色固体(175mg,78%)。
核磁:1hnmr(400mhz,c6d6)δ3.46(t,j=10.9hz,2h),3.07(dt,j=14.7,7.3hz,2h),2.26–2.13(m,2h),2.07(s,3h),1.74–1.63(m,8h),1.51(m3h),1.41–1.28(m,8h),1.26–1.10(m,11h).13cnmr(101mhz,c6d6)δ232.06,227.14,58.59(t,j=4.5hz),48.34,28.31(t,j=10.2hz),25.71(t,j=8.1hz),24.79(t,j=5.3hz),20.72,19.31,19.21,18.85.31pnmr(162mhz,c6d6)δ77.82(s).
高分辨:hr-ms(esi)calledforc19h39mnno2p2[m]+:430.1831;found:430.1834.
红外:ir:2907,2871,1900,1807,1458,1053,1021,925,816,739,686,652,621cm-1.
x-射线单晶结构如图3所示。
由上述分析结果可知所制备的目标锰络合物结构正确。
按照与实施例2相同的方法制备得到了式1、式2、式3、式4、式5、式6、式8和式10所示钴络合物。
实施例3、合成pnp-mn(co)2络合物(式11所示)
在氩气氛围下,往10ml的schlenk瓶中,先后加入pnpipr-mn(co)2br(0.15g,0.3mmol),乙醇钠(61mg,0.9mmol)和无水无氧thf(4ml)。反应室温搅拌2小时,随着反应的进行,溶液逐渐由黄色变为红色,反应结束后,抽干溶剂。接着往剩余物中加入5ml无水无氧正己烷,氩气过滤,所得滤液抽干,即为氨基锰络合物式11(87mg,69%)。
核磁:1hnmr(400mhz,c6d6)δ2.96(s,1h),1.99(d,j=4.7hz,1h),1.38(s,1h),0.98(d,j=5.7hz,3h),0.84(d,j=4.2hz,3h).13cnmr(101mhz,c6d6)δ64.89(j=10.1hz,),26.29(j=10.1hz),22.85(j=5.6hz),18.08,17.48.31pnmr(162mhz,c6d6)δ112.99(s).
x-射线单晶结构如图4所示。
由上述分析结果可知所制备的目标锰络合物结构正确。
实施例4、合成pnp-mn(co)2oac络合物(式15所示)
在氩气氛围下,往10ml的schlenk瓶中,先后加入pnpipr-mn(co)2(0.1mmol,41.4mg),无水无氧正己烷(2ml)和醋酸(0.3mmol,18mg)。反应室温搅拌0.5小时,之后将混合液放入-20度冰箱过夜,将上层正己烷溶液吸走,得浅黄色晶体(36mg,75%)。
核磁:1hnmr(400mhz,c6d6)δ8.16(s,1h),3.57(s,3h),2.95(s,2h),2.28–1.56(m,11h),1.52–0.64(m,24h).13cnmr(101mhz,c6d6)δ67.45,52.44,31.58,27.42,26.46,25.44,24.30,22.67,19.95(d,j=24.3hz),18.14(d,j=59.4hz).13.96.31pnmr(162mhz,c6d6)δ87.68(s).
x-射线单晶结构如图5所示。
由上述分析结果可知所制备的目标锰络合物结构正确。
按照与实施例1相同的方法制备得到了式12和式16所示锰络合物.
实施例5、合成顺式pnpipr-mn(co)2h络合物(式13所示)
在手套箱内,将氨基锰络合物式11(0.01mmol,4.1mg)和甲苯(1ml)加入到含针头的小瓶内,之后将小瓶放入100ml高压反应釜内。釜内充10个大气压的氢气,室温反应14小时。待反应结束,将氢气缓慢释放后膦谱跟踪反应产物。
31pnmr(162mhz,toluene):109.38ppm.
由上述分析结果可知所制备的目标锰络合物结构正确。
实施例6、合成反式pnpipr-mn(co)2h络合物(式14所示)
在手套箱内,将氨基锰络合物式11(0.01mmol,4.1mg)、乙醇(0.3mmol,13.8mg)和甲苯(1ml)先后加入到含针头的小瓶内,待反应15min完全形成乙氧基锰络合物(式12)后,将小瓶放入100ml高压反应釜内。釜内充30个大气压的氢气,室温反应14小时。待反应结束,将氢气缓慢释放后膦谱跟踪反应产物。
31pnmr(162mhz,toluene):106.86ppm.
由上述分析结果可知所制备的目标锰络合物结构正确。
实施例7、不同锰催化剂对guerbet反应结果的研究[a]
表1不同锰催化剂效果比较
[a]反应条件:(6ml,100mmol)乙醇,(6mmol)乙醇钠,(0.01mol%)催化剂在25ml的耐压釜中反应24h.[b]总的转化率为乙醇转化为高级醇的量,产率为丁醇的产率(以联苯作为内标gc定量).[c]选择性为丁醇的物质的量在高级醇类产物中所占的物质的量比.[d]ton为每mmol的催化剂可以转化乙醇到高级醇类产物的总量(mmol).[e]巴豆醇的选择性为21%.
在氩气的保护下,将锰催化剂(0.01mmol,0.01mol%)、乙醇钠(6mmol,408.3mg,6mol%)和乙醇(100mmol,6ml)先后加入到含磁力搅拌子的25ml耐压釜中,在所给温度下反应24h。反应结束后,用冰水冷却,缓慢释放出釜体内部产生的气体,向反应体系中加入联苯作为内标,用thf稀释,通过gc定量得出产物丁醇的产率及其选择性。
通过对催化剂的筛选结果可以发现,使用位阻较小的环己基和异丙基pnp类型pincer[mn](+1)催化剂(entry1-7)对乙醇有着较好的转化率以及产率;对于位阻较大的叔丁基pnp催化剂或者配位能力较弱的nnp催化剂以及[mn](+2)催化剂均未能顺利地使乙醇发生转化(entry7-11)。
实施例8、不同碱以及其它反应条件对锰催化guerbet反应结果的研究[a]
表2条件优化
[a]反应条件:(6ml,100mmol)乙醇,(6-12mmol)乙醇钠,0.01mol%[mn]-式8在25mll的耐压釜中160℃反应12-168h.转化率,产率,选择性以及ton与实例1中计算方法一致.[b]额外加入10mmol的水.[c]反应温度为180℃.[d]0.02mol%[mn]-式8.[e]0.1mol%[mn]-式8.[f](12ml,200mmol)乙醇,(12mmol)乙醇钠.[g]60ml反应釜,(0.0025mmol)[mn]-式8,(30ml,510mmol)乙醇,(30mmol)乙醇钠.[h]60ml反应釜,(0.00125mmol)[mn]-式8,(45ml,765mmol)乙醇,(46mmol)乙醇钠.[i]100ml反应釜,(0.00125mmol)[mn]-式8,(75ml,1275mmol)乙醇,(76mmol)乙醇钠.[j]巴豆醇的选择性为11%.[i]100ml反应釜,(0.00125mmol)[mn]-式8,(75ml,1275mmol)乙醇,(150mmol)乙醇钠.
通过对以上反应进行条件优化发现,使用[mn](式8)为催化剂,对碱进行筛选(entry1-5),当使用强碱时都具有较好的反应结果,当时用弱碱(如醋酸钠)时,该反应虽然具有较好的选择性(entry5),但其转化率和产率较差;增加碱的用量以及催化剂的用量,其转化率以及产率能够得到进一步的提高(entry10-12);同时还发现,对于该反应在反应装置相同时,增加其底物乙醇的用量但其它反应条件相同时,对于产物乙醇的选择性能够随之增大(entry1,13),可认为,由于反应釜可容纳气体的相对空间较少时,原位生成的氢气压力就会增大,这样会加快反应中间体的氢化速率,从而阻止了其向高级醇的转化(通过对反应体系中产生的气体进行gc检测,结果显示其气相中氢气的含量达到了81%)。最后,对反应条件进行一系列的优化后,该反应可得到高达114120的ton(entry19),其中前12h的tof为3078h-1。
实施例9、毒化实验反应研究
表3配体毒化结果比较
在氩气的保护下,将催化剂[mn](式8)(0.01mmol,0.01mol%)、乙醇钠(408.3mg,6mmol,6mol%)、乙醇(100mmol,6ml)和相应量的hg或pme3先后加入到含磁力搅拌子的25ml耐压釜中,在160℃反应24h。反应结束后,用冰水冷却,缓慢释放出釜体内部产生的气体,向反应体系中加入联苯作为内标,用thf稀释,通过gc定量得出产物丁醇的产率及其选择性。
以上结果表明,加入毒化试剂hg或者pme3对整个催化反应的转化率和产率并未有任何不利的影响,说明该催化反应是一个均相反应的过程。
实施例10、n-me保护的锰催化剂在乙醇转化过程中的研究
表4n-me催化剂结果比较
在氩气的保护下,将催化剂[mn]式9或式10(0.01mmol,0.01mol%)、乙醇钠(408.3mg,6mmol,6mol%)和乙醇(100mmol,6ml)先后加入到含磁力搅拌子的25ml耐压釜中,在160℃下反应24h。反应结束后,用冰水冷却,缓慢释放出釜体内部产生的气体,向反应体系中加入联苯作为内标,用thf稀释,通过gc定量得出产物丁醇的产率及其选择性。
从表4结果中来看,将n-h结构变为n-me之后,对于乙醇的转化产生了极大的影响。其转化率,产率以及选择性都有了非常明显的下降,因此,配体中n-h结构的存在对于乙醇到丁醇的整个转化起着非常重要的作用。
实施例11、锰催化乙醛和巴豆醛的缩合反应研究
在氩气的保护下,将锰催化剂式8(0.01mmol)(或不加入催化剂)、乙醇钠(1mmol)、乙醛或巴豆醛(1ml)先后加入到含磁力搅拌子的25ml耐压釜中,在160℃下反应4h。反应结束后,用冰水冷却,缓慢释放出釜体内部产生的气体,向反应体系中加入联苯作为内标,用thf稀释,通过gc定量得出原料缩合产物的选择性以及产率。
从上述所得结果来看,将加入催化剂与不加入催化剂对乙醛或者巴豆醛的缩合产物进行分析,其c4的选择性均并未有明显的差别,即催化剂的引入并未能提高c4的选择性从而抑制c4+产物的生成。因此,我们推测,对于乙醇的转化反应,其产物丁醇的高选择性是由于缩合中间体巴豆醛能够高效的发生氢化反应所引起的。
实施例12、锰催化乙醛和巴豆醛的氢化反应研究
在氩气的保护下,将锰催化剂-式8或9(0.01mmol)、乙醇钠(0.12mmol)、乙醛或巴豆醛(2mmol)先后加入到含磁力搅拌子的25ml高压釜中,以thf为溶剂,用惰性气体氩气置换三次高压釜中的气体之后充入10barh2,在160℃下反应24h。反应结束后,用冰水冷却,缓慢释放出釜体内部氢气,向反应体系中加入联苯作为内标,用thf稀释,通过gc定量得出氢化产物的选择性以及产率。
从上述氢化结果来看,[mn]-式8的加入对于乙醛或者巴豆醛的c4产物有着比较明显的促进作用,其c4产物的选择性得到明显提高,该结果再次说明了乙醛和氢气是乙醇向丁醇转化过程的中间体;另外,使用n-me保护的锰催化剂式9,无论对于乙醛或巴豆醛的氢化反应来说,都得到较低的c4选择性。因此,这也进一步说明含有n-h结构的催化剂其金属与配体协同发生氢转移的方式对于其反应中间体巴豆醛的氢化是至关重要的。
由上述实施例可以看出,本发明提供了一类新型的锰催化乙醇缩合生成丁醇的反应,该反应表现出了很高的催化活性(ton=114120,其中前12h的tof为3078h-1)和92%的丁醇选择性,具有很高的研究价值和应用前景。
在此说明书中,本发明已经参照特定的实施实例做了描述。但是,很显然仍可以作出各种修改和变换而不背离本发明的精神和范围。因此,说明书和附图应被认为是说明性的而非限制性的。
- 还没有人留言评论。精彩留言会获得点赞!