一种用于检测基因组目标区域的引物组合物及其应用的制作方法
本发明属于分子生物学领域,涉及一种用于检测基因组目标区域的引物组合物及其应用,具体涉及通过血浆游离dna和数字pcr检测肺癌的egfr突变位点的引物组合物,其组成的检测试剂盒及应用。
背景技术:
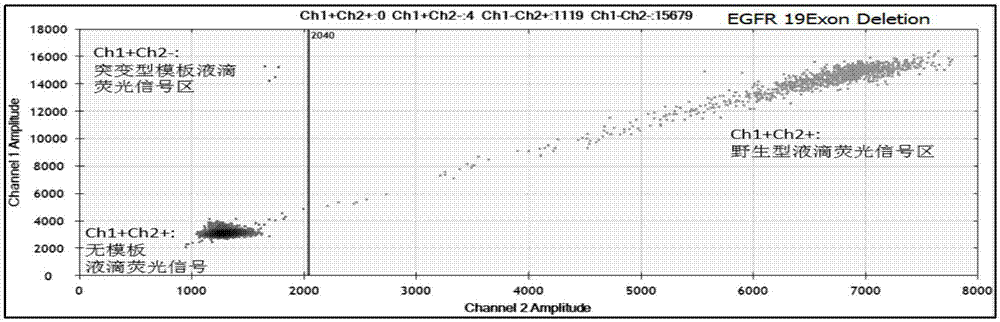
:肿瘤是一种多因素参与、多步骤发展的全身性、系统性疾病,癌症相关基因异常是导致肿瘤细胞出现逃避凋亡、无限复制、血管生成、侵袭及转移和免疫逃逸等异常生物学行为的根本原因。肿瘤靶向药物特异性作用于特定突变位点,通过抑制肿瘤细胞生长,增殖,分化抑制肿瘤进一步恶化,杀伤力强,副作用小;缺点是治疗中易出现耐药突变,因此该类药物使用时需要对靶点突变及耐药突变进行准确检测。表皮生长因子受体(epidermalgrowthfactorreceptor,egfr)表达于上皮细胞表面,在一系列人类恶性肿瘤中表现为过表达。egfr基因变异与酪氨酸激酶抑制剂tkis之间存在着显著关联,最常见的egfr变异导致21外显子第858位氨基酸由亮氨酸变为精氨酸(l858r见于40%的患者)和19外显子框架内缺失(见于45%的患者)。二者都会导致酪氨酸激酶结构域活化,且都与肿瘤对小分子tkis(厄洛替尼、吉非替尼、阿法替尼)的敏感相关。fda批准阿法替尼用于敏感egfr突变的转移性nsclc非鳞癌患者一线治疗,但t790m突变可导致tki类药物耐药,有关报道显示该突变类型见于约50%的肿瘤初始位于厄洛替尼有效后进展的患者。近些年,出现了许多基于二代或三代pcr的新技术,逐步实现使对肿瘤患者准确灵敏快速的无创检测。二代pcr技术-实时荧光定量pcr技术发明之后,可以利用荧光信号强度记录反应dna模板量,通过计算可以实现对模板起始量间接定量。arms方法基于qpcr平台检测单碱基突变,即将引物3’端最后一位碱基设计为与突变型模板互补,特异性扩增突变型模板,而野生型模板不进行扩增或被酶切,通过核酸荧光染料发出荧光信号反映突变型模板量,检测样本中是否含有突变型模板,检测下限为1%;superarms方法引物设计采用相同原理,区别是不再采用染料法,而是使用多种探针标记不同的扩增片段,提高了特异性,因此能够在单反应中加入多种引物和探针而不相互影响,通过能采集4种荧光信号的qpcr仪一次记录4种模板的荧光信号,从而检测四种单碱基突变或插入缺失突变,检测下限为0.1%,但是由于未检测野生型模板,无法对突变频率进行精确计算,仅能做定性检测。但对于二代pcr检测方法而言,其缺点在于,只能进行定性研究,无法定量检测,不能给出基因频率。数字pcr作为第三代pcr技术兴起于21世纪早期,能够将单反应体系隔离成2万个独立的液滴反应系统,每个液滴独立扩增,提高了突变序列的相对含量,降低了野生型的干扰,提高稀有变异检测的灵敏度。同时数字pcr无需标准曲线,能够绝对定量,更适用于低起始量dna的低频突变检测。血浆游离dna平均长度为170bp,其中肿瘤细胞坏死释放的dna碎片低于150bp,对于片段化的血浆dna模板,扩增子长度越短对模板的覆盖率越高,目前液体活检基于二代或三代pcr的低频突变检测方法扩增子长度在70-130bp以上,覆盖率仅23.5%-58.8%,而上下游引物加探针序列总长约50-60bp,扩增子更长的原因是序列内存在高gc区域,给引物探针设计带来困难,为了避开cg不平衡区域就必须延长扩增子长度。cn103923973a公开了一种基于数字pcr平台检测egfr第19号外显子缺失突变的方法,探针和引物设计的原理是:采用肽核酸(peptidenucleicacids,pna)锁定野生型的检测位点区域,以两对引物对、一对探针分别检测未被pna锁定的突变型和总dna模板,从而计算出突变率,此专利要求pna特异性好,反应试剂成本高。cn103911427a公开了一种基于数字pcr平台检测egfr20(t790m),egfr21(l858r)基因点突变的方法和试剂盒,检测内容非常有限,一对探针检测一种点突变类型,远远不能满足实际检测需要。使用扩增阻滞突变系统(amplificationrefracttorymutationsystem,arms),对突变靶序列进行pcr扩增,taqman探针对扩增产物进行特异性位点检测,该方法存在缺陷:arms技术引物最后一个碱基不匹配并不能完全阻滞野生型dna的扩增,所以这种方法存在假阳性的风险。基于上述问题,如何设计引物和探针,能够尽可能缩短扩增产物的长度,提高液体活检片段化模板的覆盖率,提高扩增效率,改善对低拷贝突变型模板的检出能力就成为亟待解决的问题。技术实现要素:针对现有技术的不足,本发明提供了一种用于检测基因组目标区域的引物组合物及其应用,本发明引物组合物基于数字pcr平台对血液游离dna的egfr低频突变进行检测,避免ngs测序诸多中间步骤,最大程度保留原始dna信息,所述检测试剂盒可用于检测肺癌患者egfr基因上的热点突变。第一方面,本发明提供了一种用于检测基因组目标区域的引物组合物,其包含:第一组引物,所述第一组引物包含第一正向引物和第一反向引物,其中,所述第一正向引物的3’端包含目标区域特异性正向引物,5’端包含第一核酸序列;所述第一反向引物的3’端包含目标区域特异性反向引物,5’端包含第二核酸序列;第二组引物,所述第二组引物包含第二正向引物和第二反向引物,其中,所述第二正向引物包含第一核酸序列;所述第二反向引物包含第二核酸序列;其中,所述第一核酸序列和所述第二核酸序列不同。本发明中,所述引物组合物的设计按照引物设计的规则,尽量避免二聚体、发卡结构,对每个突变位点设计所述引物组合物,即调和型双引物,即每个位点设计两对引物,第一组引物的3’端序列与模板互补,5’端添加人工序列(即第一、第二核酸序列),第二组引物与第一组引物具有相同的人工序列,同时包含第一组引物基因组目标区域互补序列的5’端部分;两对引物对应的基因组区域相同,两次扩增降低了非特异性扩增的概率,提高特异性,第二对引物的结构优化也提高了扩增效率,用于扩增非保守序列或低起始量的大片段模板。本发明中,对于血液游离dna单点突变检测,pcr产物越小模板利用率越高,而本发明通过上述设计的引物可以调节gc分布不均匀的问题,提高pcr效率,同时解决模板利用率低的问题。根据本发明,所述第一、第二核酸序列需根据引物序列的不同特异性设计,所述第一、第二核酸序列的长度分别不超过所述目标区域特异性正向引物、反向引物的50%。根据本发明,所述第二正向引物的3’端包含所述目标区域特异性正向引物5’端部分序列;所述第二反向引物的3’端包含所述目标区域特异性反向引物5’端部分序列,所述第二正向引物和第二反向引物尽量避开第一组引物的3’端的高gc区。根据本发明,所述第二组引物的正向、反向引物的总长度不超过第一组引物的正向、反向引物的90%。根据本发明,所述第一组引物与所述第二组引物的tm值相当,优选为相差不超过5℃,例如可以是5℃、4℃、3℃、2℃或1℃,进一步优选为相差不超过2℃。根据本发明,所述第一、第二组引物的tm值分别为45-65℃,例如可以是45℃、46℃、47℃、48℃、49℃、50℃、51℃、52℃、53℃、54℃、55℃、56℃、57℃、58℃、59℃、60℃、61℃、62℃、63℃、64℃或65℃,优选为55-62℃。本发明中,所述第一、第二组引物的tm值低于所述检测探针的tm值,相差不小于5℃,例如可以是5℃、6℃、7℃、8℃、9℃、10℃、11℃、12℃、15℃、18℃或20℃。根据本发明,所述第一组引物的3’端目标区域特异性引物的长度本领域技术人员可以根据需要进行调整,调整的第一正向引物、第一反向引物的tm值要满足设定的tm值即可,本申请中的第一正向引物、第一反向引物的长度分别为14-40bp,例如可以是14bp、15bp、16bp、17bp、18bp、19bp、20bp、21bp、22bp、23bp、24bp、25bp、26bp、27bp、28bp、29bp、30bp、31bp、32bp、33bp、34bp、35bp、36bp、37bp、38bp、39bp或40bp,优选为18-30bp。优选地,所述第二组引物的3’端目标区域特异性引物的长度本领域技术人员可以根据需要进行调整,调整的第二正向引物、第二反向引物的tm值要满足设定的tm值即可,本申请中的第二正向引物、第二反向引物的长度分别为14-40bp,例如可以是14bp、15bp、16bp、17bp、18bp、19bp、20bp、21bp、22bp、23bp、24bp、25bp、26bp、27bp、28bp、29bp、30bp、31bp、32bp、33bp、34bp、35bp、36bp、37bp、38bp、39bp或40bp,优选为18-30bp。本发明中,由于人工序列,即第一、第二核酸需根据引物序列的不同特异性设计,长度一般不超过第一组引物的模板互补区,人工序列的设计优选at与cg间隔分布,目的是为了优化第二组引物的结构,上述第一、第二组引物的3’端目标区域特异性引物为不包括第一、第二核酸的第一正向引物、第一反向引物、第二正向引物和第二反向引物的长度。根据本发明,所述引物组更适于短扩增子长度,以及高gc区域设计引物,扩增子的长度为不小于30bp,所述扩增子为所述引物组扩增得到的扩增产物。根据本发明,所述引物组合物用于检测egfr基因第19-21号外显子突变;优选地,所述引物组包括3组引物组或与其具有至少80%同一性,优选至少90%同一性的核苷酸序列,所述引物组的核苷酸序列如下:(1)用于检测egfr基因第19号外显子突变的引物组如下:第一正向引物如seqidno.1所示的核苷酸序列;第一反向引物如seqidno.2所示的核苷酸序列;第二正向引物如seqidno.3所示的核苷酸序列;第二反向引物如seqidno.4所示的核苷酸序列;(2)用于检测egfr基因第20号外显子突变的引物组如下:第一正向引物如seqidno.5所示的核苷酸序列;第一反向引物如seqidno.6所示的核苷酸序列;第二正向引物如seqidno.7所示的核苷酸序列;第二反向引物如seqidno.8所示的核苷酸序列;(3)用于检测egfr基因第21号外显子突变的引物组如下:第一正向引物如seqidno.9所示的核苷酸序列;第一反向引物如seqidno.10所示的核苷酸序列;第二正向引物如seqidno.11所示的核苷酸序列;第二反向引物如seqidno.12所示的核苷酸序列;所述seqidno.1-12所示的核苷酸序列如下表所示:本发明中,所述人工序列,即第一、第二核酸根据egfr基因第19-21号外显子进行特异性设计,用于检测egfr基因第19号外显子突变的第一核酸序列和第二核酸序列如seqidno.19-20所示:第一核酸序列(seqidno.19):agact;第二核酸序列(seqidno.20):ctca;用于检测egfr基因第20号外显子突变的第一核酸序列和第二核酸序列如seqidno.21-22所示:第一核酸序列(seqidno.21):ggtgcagga;第二核酸序列(seqidno.22):cacacac;用于检测egfr基因第21号外显子突变的第一核酸序列和第二核酸序列如seqidno.23-24所示:第一核酸序列(seqidno.23):cactgact;第二核酸序列(seqidno.24):ctcaga.根据本发明,所述用于检测egfr基因第19-21号外显子突变的引物组的扩增子长度为49-88bp,所述扩增子为所述引物组扩增得到的扩增产物。第二方面,本发明提供一种用于检测基因组目标区域的检测探针,所述检测探针包括野生型探针和/或突变型探针;根据本发明,所述野生型探针和所述突变型探针5’端都修饰有荧光基团,所述野生型探针和所述突变型探针5’端的荧光集团不同。根据本发明,所述荧光探针5’端修饰的荧光基团为fam、vic、hex、cy5或cy3中的任意一种或至少两种的组合。根据本发明,所述荧光探针3’端修饰的淬灭基团为bhq1、bhq2、mgb、tamra、dabcyl或eclipse中的任意一种或至少两种的组合。本发明中,使用不同荧光基团结合mgb探针标记突变型和野生型模板,对单碱基突变设计两种荧光探针分辨snp突变型模板和野生型模板,使含有突变型模板的液滴发出fam荧光,野生型模板的液滴发出vic荧光;对缺失型突变设计保守区探针和缺失区探针,含有野生型模板的液滴发出fam和vic两种荧光信号,缺失型模板的液滴仅发出fam信号。优选地,所述检测探针用于检测egfr基因第19-21号外显子突变;优选地,所述用于egfr基因第19-21号外显子突变的检测探针包括3组检测探针或与其具有至少80%同一性,优选至少90%同一性的核苷酸序列,所述检测探针的核苷酸序列如下:(1)用于检测egfr基因第19号外显子突变的检测探针如下:野生型探针的核苷酸序列如seqidno.13所示;突变型探针的核苷酸序列如seqidno.14所示;(2)用于检测egfr基因第20号外显子突变的检测探针如下:野生型探针的核苷酸序列如seqidno.15所示;突变型探针的核苷酸序列如seqidno.16所示;(3)用于检测egfr基因第21号外显子突变的检测探针如下:野生型探针的核苷酸序列如seqidno.17所示;突变型探针的核苷酸序列如seqidno.18所示。所述seqidno.13-18所示的核苷酸序列如下表所示:其中,mt表示突变型,wt表示野生型,加粗字母表示突变位点。第三方面,本发明提供一种用于检测基因组目标区域的试剂盒,其包括如第一方面所述的引物组合物。根据本发明,所述试剂盒还包括如第二方面所述的检测探针。第四方面,本发明提供一种如第一方面所述的引物组合物用于制备检测肺癌的试剂。根据本发明,所述试剂还包括如第二方面所述的检测探针。第五方面,本发明提供一种检测基因组目标区域的方法,采用如第三方面所述的检测试剂盒,包括如下步骤:1)配制pcr混合液;2)将配制的pcr混合液进行数字pcr扩增;3)将pcr扩增得到的数据进行信息处理。根据本发明,步骤1)所述的pcr混合液包括待测目标片段、引物组和pcr预混液,优选包括待测目标片段、引物组、检测探针和pcr预混液。根据本发明,所述待测目标片段为通过临床样本提取获得。根据本发明,所述临床样本为血液游离dna、手术切除组织、石蜡包埋组织切片、穿刺组织、胸水、口腔黏膜、胸腔积液、血浆或血清中的任意一种或至少两种的组合,优选为血液游离dna。根据本发明,所述引物组为如第一方面所述的引物组合物。根据本发明,所述检测探针为如第二方面所述的检测探针。根据本发明,所述pcr混合液的每个反应体系包含:优选为1-66ng的待测目标片段,含量分别为400-600nm、优选为500nm的引物组中的各条引物和含量为200-800nm、优选为250-750nm的检测探针。所述待测目标片段例如可以是1ng、2ng、3ng、5ng、6ng、8ng、10ng、12ng、15ng、16ng、18ng、20ng、22ng、25ng、26ng、28ng、30ng、32ng、35ng、36ng、38ng、40ng、42ng、45ng、46ng、48ng、50ng、52ng、54ng、56ng、58ng、60ng、62ng、64ng、65ng或66ng。所述引物组含量为400nm、410nm、420nm、430nm、440nm、450nm、460nm、480nm、490nm、500nm、510nm、520nm、530nm、540nm、550nm、560nm、570nm、580nm、590nm或600nm。所述检测探针的含量为200nm、210nm、230nm、250nm、280nm、300nm、310nm、350nm、380nm、400nm、410nm、450nm、480nm、500nm、510nm、530nm、550nm、580nm、600nm、610nm、630nm、650nm、680nm、700nm、710nm、730nm、750nm、780nm或800nm。本发明中,所述反应体系还包括相应的酶和缓冲液,所述酶和缓冲液只要能够完成pcr反应的酶和缓冲液都是可行的,在此不作特殊限定,本领域技术人员可以根据需要进行选择,本申请采用bio-rad提供的酶和缓冲液。根据本发明,步骤2)所述的pcr扩增为数字pcr扩增,所述数字pcr扩增为将步骤1)所述的pcr混合液制作为pcr微反应液滴,再进行pcr扩增。根据本发明,所述pcr扩增反应的反应条件为梯度退火温度pcr,所述梯度退火温度可以根据pcr引物的tm值进行调节,在此不作特殊限定,本发明所述梯度退火温度pcr为:对于设计好的引物tm值范围,前2-4个循环退火温度设置为所述引物tm温度上限+(1-6)℃,中间3-6循环退火温度设置为所述引物tm温度上限+(1-3)℃,后30-35循环退火温度设置为所述引物tm下限温度。本发明中,通过前几个温度较高的循环退火温度,可实现高特异性扩增,但产量较低,后续的提高循环退火温度,再对前几个循环的扩增产物进行扩增,提高产量,增强信号强度。根据本发明,对于检测egfr基因第19-21号外显子突变的引物,对于引物tm值56-59℃范围,所述梯度退火温度pcr具体程序:优选地,所述pcr扩增反应的反应条件为:92-96℃预变性3-15分钟;92-96℃变性5-50秒,60-65℃延伸30-90秒,共进行2-4个循环;92-96℃变性5-50秒,58-63℃延伸30-90秒,共进行3-6个循环;92-96℃变性5-50秒,52-60℃延伸30-90秒,共进行30-35个循环;92-96℃变性3-15分钟,2-10℃终止反应;优选地,所述pcr扩增反应的反应条件为:94-96℃预变性5-10分钟;94-96℃变性15-30秒,63-65℃延伸60-80秒,共进行2-3个循环;94-96℃变性15-30秒,60-63℃延伸60-80秒,共进行3-5个循环;94-96℃变性15-30秒,54-58℃延伸60-80秒,共进行32-35个循环;94-96℃变性5-10分钟,2-10℃终止反应;优选地,所述pcr扩增反应的反应条件为:95℃预变性10分钟;95℃变性30秒,64℃延伸60秒,共进行2个循环;95℃变性30秒,62℃延伸60秒,共进行4个循环;95℃变性30秒,56℃延伸60秒,共进行34个循环;98℃变性10分钟,4℃终止反应;根据本发明,所述pcr混合液制作为pcr微反应液滴具体包括如下:将所述pcr混合液加入微滴发生器中,生成10000-20000,优选为15000-20000个微反应液滴。根据本发明,步骤3)所述的结果为收集pcr扩增产物的信号,优选为荧光信号。根据本发明,所述信息处理具体包括:将收集的信号进行数据分析,划定阈值并统计突变频率。根据本发明,所述信息处理具体包括:将收集的荧光信号采用quantasoft(bio-rad)软件进行数据分析,划定阈值并统计突变频率。本发明中,所述结果判定,本领域技术人员可以根据阴性和已知频率的阳性标准品测试结果划定阈值,优选检测频率最接近已知频率的阈值。但并非唯一的阈值判定方法,本发明的结果判定具体为:对于19外显子缺失,当ch1+和ch2-大于0,则判定为阳性;当ch1+和ch2-为0,则判定为阴性;对于t790m位点,当ch1+和ch2-大于1,则判定为阳性;当ch1+和ch2+不大于3且ch1+和ch2-不大于1,则判定为阴性;对于l858r位点,当ch1+和ch2-大于0,则判定为阳性;当ch1+和ch2+不大于1且ch1+和ch2-为0,则判定为阴性。与现有技术相比,本发明具有如下有益效果:(1)本发明试剂盒中的引物组合物对应相同的模板区域,在同一扩增反应中发挥作用,能够保证扩增特异性前提下提高扩增效率,适用于扩增低拷贝数的短片段模板,且所述引物组合物具有高灵敏性,能够检测egfr基因23种热点低频突变,需要样本量低,适用于血浆等液体样本检测效果好,还可兼顾完整基因;(2)本发明与数字pcr其他检测试剂盒相比,极大改善了模板覆盖度,提高对目的模板的检出能力,能够使用较少的dna量检出低频突变;(3)本发明方法利用体液检测,样本采集方便,操作简便,检测周期短,检测结果直观易读,节约成本,可应用于肺癌等恶性肿瘤的临床用药指导及疗效动态监测。附图说明图1为本发明引物组合物的设计图;图2是本发明实施例2中egfr19外显子缺失的检测结果,横坐标为channel2amplitude(通道2振幅),vic通道,阈值为2040;纵坐标为channel1amplitude(通道1振幅),fam通道;图3是本发明实施例2中egfrp.t790m的检测结果,横坐标为channel2amplitude(通道2振幅),vic通道,阈值为8100;纵坐标为channel1amplitude(通道1振幅),fam通道,阈值为12700;图4是本发明实施例2中egfrp.l858r的检测结果,横坐标为channel2amplitude(通道2振幅),vic通道,阈值为2000;纵坐标为channel1amplitude(通道1振幅),fam通道,阈值为3800;图5是本发明实施例3中dna模板打断后片段检测峰图;图6(a)是本发明实施例3中egfr19外显子缺失的检测结果,;图6(b)是本发明实施例3中egfrp.t790m的检测结果,;图6(c)是本发明实施例3中egfrp.l858r的检测结果,其中,横坐标为channel2amplitude(通道2振幅),纵坐标为channel1amplitude(通道1振幅),圆圈内信号点代表突变型信号点;图7是本发明实施例4中dna模板片段长度检测峰图;图8(a)为实施例4中egfr19del阳性样本检测结果,图8(b)为实施例4中egfr19del阳性样本bio-rad检测结果,图8(c)为实施例4中egfr19del阴性样本检测结果,图8(d)为实施例4中egfr19del阴性样本bio-rad检测结果,其中,横坐标为channel2amplitude(通道2振幅),纵坐标为channel1amplitude(通道1振幅),圆圈内信号点代表突变型信号点;图9(a)为实施例4中egfrt790m阳性样本检测结果,图9(b)为实施例4中egfrt790m阳性样本bio-rad检测结果,图9(c)为实施例4中egfrt790m阴性样本检测结果,图9(d)为实施例4中egfrt790m阴性样本bio-rad检测结果,其中,横坐标为channel2amplitude(通道2振幅),纵坐标为channel1amplitude(通道1振幅),圆圈内信号点代表突变型信号点;图10(a)为实施例4中egfrl858r阳性样本检测结果,图10(b)为实施例4中egfrl858r阳性样本bio-rad检测结果,图10(c)为实施例4中egfrl858r阴性样本检测结果,图10(d)为实施例4中egfrl858r阴性样本bio-rad检测结果,其中,横坐标为channel2amplitude(通道2振幅),纵坐标为channel1amplitude(通道1振幅),圆圈内信号点代表突变型信号点;图11是本发明实施例2中egfr基因19外显子缺失阳性样本箱型图,其中,bio-rad_mt,bio-rad_wt,bio-rad_total分别代表bio-rad试剂盒检测到的突变型拷贝,野生型拷贝和总阳性拷贝数(突变型拷贝和野生型拷贝数之和),mt,wt,total分别代表试本方法检测到的突变型拷贝,野生型拷贝和总阳性拷贝数(突变型拷贝和野生型拷贝数之和);图12是本发明实施例2中egfr基因t790m阳性样本箱型图,其中,bio-rad_mt,bio-rad_wt,bio-rad_total分别代表bio-rad试剂盒检测到的突变型拷贝,野生型拷贝和总阳性拷贝数(突变型拷贝和野生型拷贝数之和),mt,wt,total分别代表试本方法检测到的突变型拷贝,野生型拷贝和总阳性拷贝数(突变型拷贝和野生型拷贝数之和);图13是本发明实施例2中egfr基因t858r阳性样本箱型图,其中,bio-rad_mt,bio-rad_wt,bio-rad_total分别代表bio-rad试剂盒检测到的突变型拷贝,野生型拷贝和总阳性拷贝数(突变型拷贝和野生型拷贝数之和),mt,wt,total分别代表试本方法检测到的突变型拷贝,野生型拷贝和总阳性拷贝数(突变型拷贝和野生型拷贝数之和)。具体实施方式下面结合附图和实施例对本发明作进一步的详细说明。可以理解的是,此处所描述的具体实施例仅仅用于解释本发明,而非对本发明的限定。另外还需要说明的是,为了便于描述,附图中仅示出了与本发明相关的部分而非全部结构。实施例1检测试剂盒的制备(1)引物组合物的制备设计引物探针在数字pcr平台检测egfr基因的23种热点突变,所述egfr基因突变位点如下表1所示:表1根据gefr的热点突变,设计引物如图1所示,具体的引物序列如下表2所示:表2(2)检测探针的制备根据gefr的热点突变,设计的检测探针如下表3所示:表3其中,mt表示突变型,wt表示野生型,加粗字母表示突变位点;(3)试剂盒的组装将所述引物组合物和检测探针与试剂盒相关的试剂进行组装,制备成所述检测试剂盒。实施例2检测egfr位点突变所述检测egfr位点突变的方法,包括如下步骤:(1)血浆分离:用抗凝管采集患者6ml新鲜血液4小时之内分离血浆,将采血管放置4℃2000g10min,轻轻取出,将上清转移至新的离心管,4℃16000g10min离心,吸取上清至新的2mlep管中,-80℃保存;(2)dna提取:采用3ml血浆,使用qiagen公司人循环dna提取试剂盒(qiaampcirculatingnucleicacidkit)按说明书步骤提取dna,将dna溶解至30μlave缓冲液或30μl分子生物水;(3)配制体系:(a)egfr19del位点的体系组成如下表4所示:表4组分终浓度2×ddpcrsupermix(nodutp)(bio-rad)11μlseqidno:1-4各500nmseqidno:13-14各250nmdna血浆dna:10μl分子生物水补足22μl总体积22μl(b)egfrt790m位点的体系组成如下表5所示:表5组分终浓度2×ddpcrsupermix(nodutp)(bio-rad)11μlseqidno:5-8各500nmseqidno:15-16各750nmdna血浆dna:10μl分子生物水补足22μl总体积22μl(c)egfrl858r位点的体系组成如下表6所示:表6组分终浓度2×ddpcrsupermix(nodutp)(bio-rad)11μlseqidno:9-12各500nmseqidno:17-18各250nmdna血浆dna:10μl分子生物水补足22μl总体积22μl(4)将配制的pcr反应体系制作为pcr微反应液滴,进行数字pcr扩增:放入美国bio-rad公司的全自动微滴发生器液中,进行液滴发生,将每个体系分成20000个液滴,金属薄膜180℃5秒热封之后之后进行数字pcr扩增,具体数字pcr扩增程序如下表7所示:表7(5)将pcr扩增得到的数据进行信息处理:使用bio-rad公司的微滴检测仪检测液滴荧光,并使用quantasoft软件进行数据分析,划定阈值并统计突变频率,检测结果二维图、阈值划定如图2-4,检测结果判定如下表8所示:表8从图2可以看出,阈值分为三个区域,ch1+ch2-区域为突变型模板液滴荧光信号区,fam信号较强,ch1+ch2+区域为野生型模板液滴荧光信号区,两种荧光信号均较强,ch1-ch2-区域液滴不包含dna模板,仅发出非激发态的荧光信号,fam和vic荧光值均较低;从图3可以看出,将阈值分为四个区域,ch1+ch2-区域为突变型模板液滴荧光信号区,fam信号较强,ch1+ch2+区域液滴同时包含突变型模板和野生型模板,两种荧光信号均较强,ch1+ch2-为野生型液滴荧光信号区,vic荧光信号较强,ch1-ch2-区域液滴不包含dna模板,仅发出非激发态的荧光信号,fam和vic荧光值均较低;从图4可以看出,阈值分为四个区域,ch1+ch2-区域为突变型模板液滴荧光信号区,fam信号较强,ch1+ch2+区域液滴同时包含突变型模板和野生型模板,两种荧光信号均较强,ch1+ch2-为野生型液滴荧光信号区,vic荧光信号较强,ch1-ch2-区域液滴不包含dna模板,仅发出非激发态的荧光信号,fam和vic荧光值均较低;综合图2-4可以看出,egfr基因各个位点检测结果二维图信号群聚集和信号群之间分离情况良好,二维信号图直观易辨。实施例3采用从horizon公司购买的egfr阳性标准品hd734与egfr阴性标准品hd709混合,利用超声方法片段化,作为模拟样本并梯度稀释不同的频率及浓度进行检测,dna模板打断后检测结果如图5所示,从图5可以看出,dna模板打断后片段与已知长度片段峰图相比,片段主峰位于200bp和300bp之间;采用实施例1中的检测试剂盒用实施例2中的方法进行检测,结果见表9-11,部分检测结果二维图如图6(a)-图6(c)所示:表9egfr基因19外显子缺失位点dna量(ng)203050801001200.04%-++0.02%+-+0.01%+++0%-表10egfr基因t790m位点dna量(ng)203050801001200.04%+++0.02%-++0.01%-++0%-表11egfr基因l858r位点dna量(ng)203050801001200.04%+-+0.02%-++0.01%---0%-注:“+”表示检出阳性,“-”表示检出阴性。从图6(a)-图(c)和表9-11的检测结果可以看出,本方法检测下限为egfr19del突变0.02%,80ng;egfrl858r突变0.01%,100ng;egfrt790m突变0.01%,80ng。实施例4使用从horizondx购买的血浆标准品hd780,其中1%突变型dna与野生型dna混合频率为0.5%模拟血浆dna,hd780系列的野生型dna作为阴性对照,分选出150bp以下的小片段模拟肿瘤细胞坏死释放的游离dna用于检测本专利检测范围的基因突变,将实施例1中的检测试剂盒与bio-rad公司的数字pcr检测试剂盒进行对比。dna模板片段打断后检测结果如图7所示,可见长度主峰在129bp。本发明方法与bio-rad检测试剂盒的扩增子长度对比如表12所示:表12本方法与bio-rad检测试剂盒的扩增子长度对比*:bio-rad扩增子长度数据来源于bio-rad官网。阳性样本每反应模板量为10ng,阴性样本每反应5ng,实验设计如表13,按实施例2中的方法进行检测,bio-rad按说明书配制体系并扩增:表13使用bio-rad公司的微滴检测仪检测液滴荧光,并使用quantasoft软件进行数据分析,划定阈值并统计突变频率。检测数据如表14,部分检测结果二维图如图8(a)-图8(d)、图9(a)-图9(d)和图10(a)-图10(d)所示,数据分布箱型图如图11-13所示:表14检出率=检测结果为阳性的样本例数/阳性样本例数;特异性=检测结果为阴性的样本例数/阴性样本例数。从图8(a)-图8(d)、图9(a)-图9(d)和图10(a)-图10(d)比较可以看出,本发明检测试剂盒与bio-rad试剂盒检测结果二维图中信号群聚集情况,突变型信号群和野生型信号群之间分离情况均较良好。根据表14、图8(a)-图8(d)、图9(a)-图9(d)和图10(a)-图10(d)进行数据统计,结果如表15所示,使用r3.4.1软件进行双尾异方差t检验差异显著性分析,结果如表16-17所示:表15数据统计结果mt:突变型拷贝数;wt:野生型拷贝数;total:总拷贝数=突变型拷贝数+野生型拷贝数;freq.:突变频率.表16检验双尾异方差分析差异显著性(阳性样本)倍数=本方法拷贝数/bio-rad拷贝数;mt:突变型拷贝数;wt:野生型拷贝数;total:总拷贝数=突变型拷贝数+野生型拷贝数;freq.:突变频率表17t检验双尾异方差分析差异显著性(阴性样本)egfr基因t790m-l858r-19exondel-wt0.0085590.0083500.043653wt判断差异极显著差异极显著差异显著wt:野生型拷贝数;表18bio-rad检测试剂盒与本发明试剂盒的比较检出率特异性拷贝数检出能力检测时间试剂成本/例本发明100%100%1.5~1.8倍140min40元bio-rad100%100%-140min198元从表15-18结果和图11-13可以看出,本发明方法与bio-rad检出率与特异性均为100%,t790m突变检测,本方法检出突变型拷贝数高于bio-rad的1.8倍,野生型拷贝数是bio-rad的1.6倍,且差异显著;l858r突变检测,本方法检出突变型拷贝数是bio-rad的1.6倍,野生型拷贝数是bio-rad的1.5倍,且差异显著,19外显子缺失突变,本方法检出突变型拷贝数是bio-rad的1.3倍,差异不显著,野生型拷贝数是bio-rad的1.6倍,差异显著推测为两者扩增片段长度较为接近导致。证明本发明方法对于碎片化dna模板,无论突变型还是野生型模板检出能力均强于同平台的其他设计方法,本方法与bio-rad检测试剂盒检测时间都需要约140min,但试剂成本(包括预混液,引物及探针)仅为bio-rad的20%。综上所述,本发明与数字pcr其他检测试剂盒相比,极大改善了模板覆盖度,提高对目的模板的检出能力,能够使用较少的dna量检出低频突变;本发明方法利用体液检测,样本采集方便,操作简便,检测周期短,检测结果直观易读,节约成本,可应用于肺癌等恶性肿瘤的临床用药指导及疗效动态监测。注意,上述仅为本发明的较佳实施例及所运用技术原理。本领域技术人员会理解,本发明不限于这里所述的特定实施例,对本领域技术人员来说能够进行各种明显的变化、重新调整和替代而不会脱离本发明的保护范围。因此,虽然通过以上实施例对本发明进行了较为详细的说明,但是本发明不仅仅限于以上实施例,在不脱离本发明构思的情况下,还可以包括更多其他等效实施例,而本发明的范围由所附的权利要求范围决定。序列表<110>天津华大医学检验所有限公司;广州华大基因医学检验所有限公司;深圳华大基因股份有限公司<120>一种用于检测基因组目标区域的引物组合物及其应用<130>2017<141>2017-09-04<160>24<170>siposequencelisting1.0<210>1<211>22<212>dna<213>人工合成序列()<400>1agactcctcacctccaccgtgc22<210>2<211>20<212>dna<213>人工合成序列()<400>2ctcagaggcagccgaagggc20<210>3<211>18<212>dna<213>人工合成序列()<400>3agactcctcacctccacc18<210>4<211>17<212>dna<213>人工合成序列()<400>4ctcagaggcagccgaag17<210>5<211>33<212>dna<213>人工合成序列()<400>5ggtgcaggagagaaagttaaaattcccgtcgct33<210>6<211>27<212>dna<213>人工合成序列()<400>6cacacacgcagaaactcacatcgagga27<210>7<211>23<212>dna<213>人工合成序列()<400>7ggtgcaggagagaaagttaaaat23<210>8<211>19<212>dna<213>人工合成序列()<400>8cacacacgcagaaactcac19<210>9<211>26<212>dna<213>人工合成序列()<400>9cactgactcaccgcagcatgtcaaga26<210>10<211>22<212>dna<213>人工合成序列()<400>10ctcagactcttccgcacccagc22<210>11<211>17<212>dna<213>人工合成序列()<400>11cactgactcaccgcagc17<210>12<211>18<212>dna<213>人工合成序列()<400>12ctcagactcttccgcacc18<210>13<211>22<212>dna<213>人工合成序列()<400>13cggagatgttgcttctcttaat22<210>14<211>20<212>dna<213>人工合成序列()<400>14aaagccaacaaggaaatcct20<210>15<211>17<212>dna<213>人工合成序列()<400>15atgagctgcgtgatgag17<210>16<211>17<212>dna<213>人工合成序列()<400>16atgagctgcatgatgag17<210>17<211>15<212>dna<213>人工合成序列()<400>17ttttgggctggccaa15<210>18<211>15<212>dna<213>人工合成序列()<400>18ttttgggcgggccaa15<210>19<211>5<212>dna<213>人工合成序列()<400>19agact5<210>20<211>4<212>dna<213>人工合成序列()<400>20ctca4<210>21<211>9<212>dna<213>人工合成序列()<400>21ggtgcagga9<210>22<211>7<212>dna<213>人工合成序列()<400>22cacacac7<210>23<211>8<212>dna<213>人工合成序列()<400>23cactgact8<210>24<211>6<212>dna<213>人工合成序列()<400>24ctcaga6当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1