一种苯并咪唑类络合物的制备方法与流程
本发明属于药物化学领域,涉及药物合成,具体涉及一种具备光学活性的苯并咪唑类络合物的制备方法。
背景技术:
苯并咪唑类化合物具有弱碱性,其对胃壁细胞膜的h+,k+-atp酶系,有抑制作用,阻止胃酸分泌,从而确定了苯并咪唑类化合物作为质子泵抑制剂的地位。
经过长期的研究发现,抑制h+/k+-atp酶的药物必须具有3个结构部分:吡啶环、so基和苯并咪唑环。目前几种上市的ppis多为苯并咪唑类衍生物,它通过对吡啶环或苯并咪唑环进行不同的修饰而增强其抑制胃酸的功能。
瑞典haessel公司在1974年和1976年分别研制出替莫拉唑(timoprazole)和匹克拉唑(picoprazole)。奥美拉唑(omeprazole)是由瑞典astra公司首创,1987年在瑞典上市,2000年奥美拉唑曾经以46亿美元的单品销售额,在世界畅销药物中排名第一,已收录为我国基本药物。埃索美拉唑是奥美拉唑的s-对映异构体,也就是奥美拉唑的左旋异构体,由阿斯利康制药公司开发。埃索美拉唑具有强烈且持久的酸抑制作用,同时对胃黏膜也有一定的保护作用,是目前治疗胃酸相关性疾病的优选药物。
兰索拉唑(lansoprazole)是有由日本武田公司开发,1992年在上市,对大鼠胃酸分泌的抑制作用是奥美拉唑的2-3倍,对幽门螺旋杆菌有较强的抑制作用,mic50为16mg/l,而奥美拉唑为64mg/l,体外实验24小时杀菌率为99.9%。研究兰索拉唑与奥美拉唑作用无明显差异。2009年1月30日美国fda批准日本武田制药公司研发的食管炎治疗新药兰索拉唑的对映体右兰索拉唑(dexlansoprazole)上市,又称右旋拉索拉唑,用于治疗与非糜乱性胃食管反流病相关的胃灼热及不同程度的糜乱性食道炎。上市剂型为缓释胶囊,可使药物分别在1-2小时和4-5小时分别出现两个需要浓度峰值,该药白天或夜间伴胃灼热症状的胃食管反流病患者产生长达24小时的抑酸作用和持续的症状缓解效果。
泮托拉唑(pantoprazole)的适应症范围与奥美拉唑基本相同,耐受性更好,由德国百克顿公司研发,1994年10月在南非上市,泮托拉唑的左旋异构体((s)-(-)-pantoprazole)由印度emcure公司于2006年开发上市。其与泮托拉唑和右旋泮托拉唑相比,具有疗效好,不良反应少,使用剂量小等优点。
由日本卫材公司开发,1997年上市的雷贝拉唑(rabeprazole)代谢途径较为特殊,主要经非酶途径代谢为雷贝拉唑硫醚,受肝脏cyp酶系影响较小,是受cyp2c19最小的质子泵抑制剂,因此用药过程更安全,药物间相互作用更小,且无明显个体差异。印度emcure医药公司研发的手性转换产物右旋雷贝拉唑(dexrabeprazole)于2007年9月首先在印度上市,现在已经在欧美上市,但我国目前尚未有本品上市,也没有进口注册的申请。右旋雷贝拉唑对胃烧灼痛及胃回流的效果比雷贝拉唑有明显的提高,并且在症状缓解时间上明显快于雷贝拉唑,右旋雷贝拉唑还有疗效确切,安全性好等特点。下式为上述几个药物的化学结构:
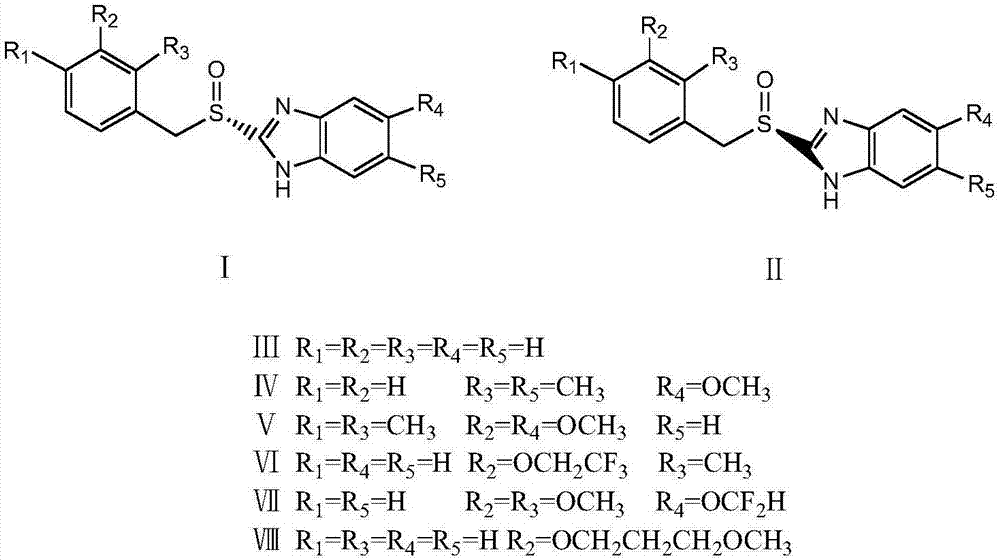
其中,式ⅲ、ⅳ、ⅴ、ⅵ、ⅶ、ⅷ分别为替莫拉唑、匹克拉唑、奥美拉唑、兰索拉唑、泮索拉唑、雷贝拉唑。
在上述苯并咪唑结构中,存在一个手性中心——硫原子,因此具有两个光学异构体,由于单一光学纯度的药物与外消旋药物相比,具有更高的生理活性和更好的药物动力学特性,可以减少药物的摄入量和减小药物对肝脏的负担,减少毒副作用的产生,也减轻了对环境的污染。如下式所示:
式ⅰ为该类化合物的(s)-异构体,式ⅱ为该类化合物的(r)-异构体。因此制药企业一直都在寻找更多的制备光学纯的单一异构体的制备方法。奥美拉唑、兰索拉唑、泮索拉唑和雷贝拉唑都有单一光学纯度的药品上市,但是现有技术的方法,将其外消旋体拆分成为单一异构体的方法很难达到光学纯度要求,或者成本较高。
技术实现要素:
为了克服现有技术的缺陷,本发明旨在是提供一种工艺简单、成本低廉的制备光学纯度高、收率高的苯并咪唑类方法。
本发明提供了一种光学纯的苯并咪唑类络合物2的制备方法,其特征在于,包括以下步骤:
(1)碱性条件下,(l)-苹果酸与苯并咪唑化合物类质子泵抑制剂形成苯并咪唑络合物1;
(2)碱性条件下,步骤(1)所述的苯并咪唑络合物1与(s)-2-羟基-2-苯乙酰氯形成苯并咪唑络合物2的粗品;
(3)将步骤(2)中所述的苯并咪唑络合物2的粗品重结晶,即得光学纯的苯并咪唑络合物2;
其中,所述的苯并咪唑化合物类质子泵抑制剂为替莫拉唑、匹克拉唑、奥美拉唑、兰索拉唑、潘妥拉唑、雷贝拉唑或泮托拉唑。
优选的,所述步骤(1)中(l)-苹果酸与苯并咪唑化合物类质子泵抑制剂的用量摩尔比为0.2:1~2:1;进一步优选的,摩尔比为0.5:1~1.2:1。
优选的,步骤(1)中使用的溶剂为甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇中的任意一种或一种以上;进一步优选的,步骤(1)中使用的溶剂为甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇中的任意两种混合物;更优选的,步骤(1)中使用的溶剂为甲醇、乙醇二者的混合物。
进一步优选的,所述两种溶剂体积比为1:2~1:15;更优选的,所述两种溶剂体积比为1:4~1:6。
优选的,步骤(1)的反应温度为0℃~50℃;进一步优选的,步骤(1)的反应温度为20℃~30℃。
优选的,步骤(1)、(2)中所用的碱性试剂为氢氧化钾、氢氧化钠、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾中的一种或一种以上;进一步优选的,步骤(1)、(2)中所用的碱性试剂为氢氧化钾和/或氢氧化钠。
进一步优选的,步骤(1)、(2)中所用的碱性试剂与苯并咪唑络合物1的摩尔比为0.8:1~2:1;更优选的,所述步骤(1)、(2)中所用的碱性试剂与苯并咪唑络合物1的摩尔比为1:1~1.2:1。
优选的,步骤(2)中,苯并咪唑络合物1与(s)-2-羟基-2-苯乙酰氯用量摩尔比为0.2:1~2:1;进一步优选的,摩尔比0.5:1~1.2:1。
优选的,步骤(2)中使用的溶剂为非质子性溶剂;进一步优选的,步骤(2)中使用的溶剂为甲苯、乙苯、二甲苯、二氯甲烷、二氯乙烷、四氢呋喃、乙酸乙酯任意一种或一种以上;更优选的,所述步骤(2)中使用的溶剂为四氢呋喃。
优选的,步骤(2)与步骤(1)中使用的溶剂量之比为1:2~1:15;优选的,步骤(2)与步骤(1)中使用的溶剂量之比为1:4~1:6。
优选的,步骤(2)的反应温度为0℃~50℃;进一步优选的,所述反应温度为20℃~30℃。
优选的,步骤(2)的反应时间为2~72h;进一步优选的,所述反应时间为2~24h;更优选的,所述反应温度为3~10h;最优选的,所述反应温度为4~8小时。
优选的,步骤(3)中重结晶使用的溶剂为甲醇、乙醇、丙醇、异丙醇、丁醇、异丁醇中的一种或一种以上;进一步优选的,重结晶使用的溶剂为乙醇和/或异丙醇。
优选的,所述步骤(3)中重结晶步骤为先升温溶解,然后再低温析晶,所述的升温溶解温度为20℃~100℃,优选40℃~80℃,更优选50℃~70℃;低温析晶温度为0℃~20℃,优选5℃~10℃。
有益效果
1.本发明给出的反应步骤和操作非常简便,降低了工艺复杂性及纯化次数,易于扩大化生产;
2.采用本发明方法制备的产品的光学异构体纯度极高,纯度≥99.7%,避免产生杂质,保证了产品质量;
3.本发明采用的制备方法避免使用强腐蚀性和挥发性溶剂,溶剂成本低廉且环保,最大程度地减少了环境污染。
附图说明
图1苯并咪唑类化合物的化学结构式;
图2苯并咪唑类化合物的r-/s-构型异构体的化学结构式;
图3制备苯并咪唑类络合物的反应路线示意图;
图4制备奥美拉唑络合物的反应路线示意图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚明了,下面结合具体实施方式并参照附图,对本发明进一步详细说明。应该理解,这些描述只是示例性的,而并非要限制本发明的范围。
预实施例:奥美拉唑络合物1的制备
将10克氢氧化钠分散在200毫升异丙醇中,室温搅拌溶清,加入50克奥美拉唑(345.42,0.145摩尔),室温搅拌4-5小时,过滤,滤饼用50毫升乙酸乙酯洗涤,干燥得53克(0.137摩尔)奥美拉唑钠。
将53克奥美拉唑钠(385.41,0.137摩尔)分散在200毫升正丙醇和5毫升水的混合溶剂中,搅拌2小时,过滤,滤液中加入500毫升丙酮,控制体系温度在35-40℃,依次加入d-酒石酸二乙酯、四异丙基氧钛、二异丙胺,搅拌30分钟,加入20克(l)-苹果酸,反应3小时后,过滤,滤饼用150毫升丙酮洗涤,干燥得49.4克奥美拉唑苯并咪唑络合物1(0.103摩尔,479.51),收率75%,异构体纯度80.5%。
实施例1:
奥美拉唑苯并咪唑络合物149.4克(0.103摩尔)分散在250毫升乙醇中,在20℃下滴加43克10%的氢氧化钠水溶液(0.115摩尔,1.04倍),搅拌2小时,减压蒸干乙醇,加入250毫升四氢呋喃,分液,有机层用20克无水硫酸钠干燥,过滤,滤液加入(s)-2-羟基-2-苯乙酰氯20.5克(170.59,0.12摩尔,1.16倍),在20℃反应6小时,蒸干四氢呋喃,慢慢加入异丙醇,并升温到65℃,溶清后停止滴加异丙醇,慢慢降温到5℃,并维持4小时,过滤,干燥得到45.2克(0.08755)奥美拉唑苯并咪唑络合物2(516.01),收率85%,异构体纯度99.8%。
实施例2
奥美拉唑苯并咪唑络合物149.4克(0.103摩尔)分散在220毫升异丙醇中,在20℃下滴加35克10%的氢氧化钠水溶液(0.115摩尔,1.04倍),搅拌3小时,减压蒸干异丙醇,加入250毫升四氢呋喃,分液,有机层用20克无水硫酸钠干燥,过滤,滤液加入(s)-2-羟基-2-苯乙酰氯20克(170.59,0.12摩尔,1.16倍),在20℃反应4小时,蒸干四氢呋喃,慢慢加入乙醇,并升温到50℃,溶清后停止滴加乙醇,慢慢降温到10℃,并维持4小时,过滤,干燥得到39.3克(0.07622)奥美拉唑苯并咪唑络合物2(516.01),收率74%,异构体纯度99.6%。
实施例3
兰索拉唑苯并咪唑络合物150克(0.135摩尔)分散在220毫升异丙醇中,在20℃下滴加35克10%的氢氧化钠水溶液,搅拌3小时,减压蒸干异丙醇,加入250毫升四氢呋喃,分液,有机层用20克无水硫酸钠干燥,过滤,滤液加入(s)-2-羟基-2-苯乙酰氯20克(170.59,0.12摩尔,0.88倍),在20℃反应4小时,蒸干四氢呋喃,慢慢加入乙醇,并升温到50℃,溶清后停止滴加乙醇,慢慢降温到10℃,并维持4小时,过滤,干燥得到38.8克兰索拉唑苯并咪唑络合物2,收率72%,异构体纯度99.4%。
实施例4
泮索拉唑苯并咪唑络合物150克(0.130摩尔,383.37)分散在220毫升异丙醇中,在20℃下滴加35克10%的氢氧化钠水溶液,搅拌3小时,减压蒸干异丙醇,加入250毫升四氢呋喃,分液,有机层用20克无水硫酸钠干燥,过滤,滤液加入(s)-2-羟基-2-苯乙酰氯20克(170.59,0.12摩尔,0.92倍),在20℃反应4小时,蒸干四氢呋喃,慢慢加入乙醇,并升温到50℃,溶清后停止滴加乙醇,慢慢降温到10℃,并维持4小时,过滤,干燥得到37.4克泮索拉唑苯并咪唑络合物2,收率75%,异构体纯度99.5%。
- 还没有人留言评论。精彩留言会获得点赞!