一种基于单引物探针捕获检测微生物目标序列的方法与流程
本发明涉及微生物检测技术领域,具体涉及一种基于单引物探针捕获检测微生物目标序列的方法。
背景技术:
随着测序技术的发展,二代测序(ngs)检测方法在各个领域中的优势逐步凸显,与传统的研究方法相比其快速省时、结果精确,可以准确灵敏地检测物种dna序列的优势被广泛应用于各个研究领域中。但由于目前全基因组测序(wgs)的周期相对较长,测序成本相对较高仍然限制了wgs的应用范围。而目标序列捕获技术的应用刚好解决了wgs测序时间长及成本高的问题,同过对特异的目标序列的捕获富集减少了测序结果中不需要的数据所占比例提高了目标序列检出比例使测序所需的数据量大大减少进而降低检测成本。尤其是对微生物的研究,不同样本类型的检测结果中都会含有不同程度的背景序列导致对测序数据量的要求较高,同是对目标微生物的检测结果分析也造成干扰。
通过捕获的方法对目标序列进行特异性检测技术包括:罗氏nimblegen序列捕获技术,该技术通过固相或液相芯片对目的片段进行捕获,探针长度为50-105bp。主要流程包括,基因组片段化及末端修复、接头连接、杂交、芯片洗脱、目的片段洗脱、扩增和测序。安捷伦sureselectdna捕获阵列技术,该技术与nimblegen技术类似,也是通过固相或液相芯片对目的片段进行捕获,探针长度为120bp左右。主要流程包括,基因组片段化及末端修复加a、接头连接、扩增、杂交、洗脱、扩增和测序。用于捕获测序的ampliseqon-demandpanels技术,通过多重pcr将目的片段进行富集,再对富集后的片段进行建库、测序。
罗氏nimblegen序列捕获技术和安捷伦sureselectdna捕获阵列技术的探针长度均较长,相较于短探针而言合成成本高。nimblegen建库流程中杂交后需要两次洗脱步骤,增加了操作时间、复杂度与样本的不必要损失,同时洗脱试剂增加了建库成本。安捷伦技术杂交和洗脱时间均较长不适合快速检测需求,同时增加了一步扩增反应,也增加了建库操作时间。ampliseq技术利用多重pcr进行富集时由于不同引物的扩增效率存在差别,以及引物间的相互影响导致不同目的片段的富集结果不均一,不适合做定量分析。pcr扩增循环数多容易引入位点突变,影响结果判断。
技术实现要素:
本发明提供一种基于单引物探针捕获检测微生物目标序列的方法,在建库流程中将接头连接在探针结合位点上游,使得后续扩增时只能对捕获到的片段进行富集,省去现有方法通过洗脱进行目的片段分离的步骤,简化操作步骤,减少目的片段不必要的损失,且杂交时间相对较短,适合快速检测需求;并且单引物探针即可进行捕获,减少前期探针设计的复杂度,而且探针长度较短合成成本较低;扩增时采用公用引物去除不同引物富集产生的不均一性。
本发明通过如下技术方案实现:
一种基于单引物探针捕获检测微生物目标序列的方法,包括:
将包含微生物目标序列的dna样本片段化,然后进行末端修复;
将末端修复后的dna片段连接接头;
使用单引物探针与靶序列的探针结合位点退火并延伸以进行靶序列捕获,其中上述探针结合位点位于上述靶序列的上游或下游,上述单引物探针包括探针序列部分和位于上述探针序列部分5’端的公共序列部分,其中上述探针序列部分用于与上述探针结合位点退火,上述公共序列部分用作通用pcr引物的结合位点;
使用一对通用pcr引物对捕获的靶序列进行扩增,得到扩增产物;对上述扩增产物进行测序分析,获得目标序列的序列信息。
上述方案中通用pcr引物可与接头序列和公共序列配对进行pcr扩增。
进一步地,上述dna样本片段化至150bp-300bp小片段。
进一步地,上述接头包含标签序列;优先地,上述接头进一步包含一段随机序列,上述随机序列与上述标签序列相邻。上述随机序列为核苷酸随机组合的序列,可用于区分每条扩增的目标序列,随机序列的长度一般为4-12nt,优先的长度为6nt。
进一步地,上述探针序列部分的长度是35-40bp。
进一步地,上述单引物探针经梯度退火与上述探针结合位点结合。
进一步地,上述梯度退火是从80℃开始梯度降温至60℃。
进一步地,上述接头的序列是seqidno:5和seqidno:6。
进一步地,上述测序平台公共接头序列部分的序列是seqidno:7。
进一步地,上述测序平台通用pcr引物的序列是seqidno:8和seqidno:9。
本发明的方法,降低了应用二代测序技术的检测成本,通过单引物探针富集目标序列降低了非目标序列的检出,提高了目标序列的检出率进而降低了测序使用的数据量使得检测成本大大降低,同时也使得对测序结果数据的分析更加容易;与pcr富集技术相比,本发明由于捕获后在片段两端连接了相同的接头使得后续pcr环节使用公共引物进行扩增,从而去除了常规用不同pcr引物扩增时所产生的引物扩增偏向性导致的检测结果不准确性;单引物探针捕获只需一条探针即可,降低了设计引物对的繁琐程度,同时也消除了多重pcr中引物交叉反应导致非特异扩增的状况,使得检测结果更加准确可靠。此外,使用bgiseq-100平台进行测序可以有效缩短测序时间实现目标序列快速检测。
附图说明
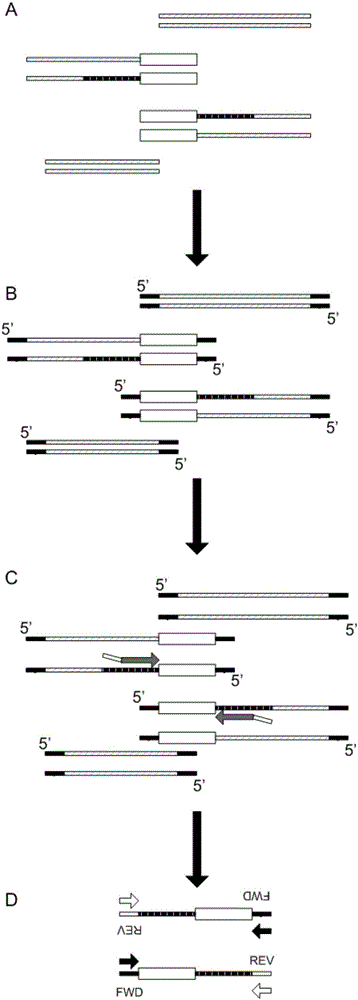
图1为本发明实施例中微生物检测文库构建原理示意图;
图2为本发明实施例中文库质量检测合格的文库的agilent2100结果图;
图3为本发明实施例中金黄色葡萄球菌4条探针的捕获结果,横坐标分别表示金黄色葡萄球菌的4条探针,纵坐标是比对上数据库金黄色葡萄球菌基因组的读长(reads)数,黑色柱子代表阳性样本,灰色柱子代表阴性对照(值较低),结果表明通过本发明实施例可以明确判定阳性样本的检出。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。在以下的实施方式中,很多细节描述是为了使得本发明能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本发明相关的一些操作并没有在说明书中显示或者描述,这是为了避免本发明的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
图1示出了本发明实施例中微生物检测文库构建原理,单引物探针捕获是在靶序列上游或下游设计一条寡核苷酸探针,在寡核苷酸探针的5’端连接一段接头的公有序列。提取待检测样本dna,样本dna片段化至150-300bp小片段,打断的dna经末端修复至平末端后连接可以区分不同样本的标签序列(barcode)接头,然后寡核苷酸探针经梯度退火与目标区域结合,通过恒温延伸来覆盖整个靶序列区域,随后经公有引物pcr扩增对捕获的序列进行富集,再通过测序平台测序,进而分析靶序列的碱基特征。
常用的高通量测序技术平台有illumina公司的hiseq系列,hiseq_2500可一次性下机最大600g碱基数据,但是周期大约需要8天左右,而bgiseq-100平台一次性产出9g碱基数据,只需要6h左右。因此,bgiseq-100平台是一个更高效的选择。因此,作为一种优选实施方式,图1示出的是基于bgiseq-100平台的单引物探针捕获流程示意图,然而,应当理解,本发明的方法也可以在其他测序平台上实现。
如图1所示,本发明实施例的方法包括:
a.将提取的样本总-dna片段化并末端修复(图1中空白框表示靶序列,断裂黑色填充框表示靶序列的探针结合位点)。
b.末端修复后片段连接接头,可以只加入含有标签序列的接头,因此片段两端被连接了相同的接头序列(图1中黑色填充框部分),使得后续未被捕获的片段不能通过pcr扩增得到富集;在其他实施方式中,接头还包括一段随机碱基,其位于标签序列前,用于区分产物是否来自同一扩增子。
c.单引物探针包括探针序列部分(图1中灰色箭头)和位于探针序列部分5’端的公共序列部分(例如测序平台公共接头序列部分,图1中灰色箭头左端的空白框),使用单引物探针与靶序列的探针结合位点退火并延伸以进行靶序列捕获。在图1中,探针结合位点位于靶序列的上游或下游,探针序列部分用于与探针结合位点退火,公共序列部分用作通用pcr引物的结合位点。该步骤中,单引物探针经梯度退火与上述探针结合位点结合,例如从80℃开始梯度降温至60℃。在优选的实施例中,探针序列部分的长度是36bp,测序平台(bgiseq-100平台)公共接头序列部分的长度是31bp,总长度较短、合成成本低。
d.延伸后产物用bgiseq-100平台通用pcr引物(图1中黑色箭头fwd和空白箭头rev)进行扩增,只有经杂交延伸后接入两端通用接头引物的片段才能被扩增,扩增产物即为构建好的dna捕获文库。然后,通过二代测序获得dna捕获文库的序列,并经分析得到靶序列的碱基序列信息。
以下通过实施例详细说明本发明的技术方案,应当理解,实施例仅是示例性的,不能理解为对本发明保护范围的限制。以下实施例所用到的试剂信息如下表1所示。
表1
实施例1
1.对金黄色葡萄球菌(sa)基因组中2个250bp的特异区域设计4条探针进行捕获,按照图1所示的文库构建流程进行文库构建。所使用的探针序列部分(图1中灰色箭头)如表2所示。
表2
2.将金黄色葡萄球菌的菌液用天根dp-302试剂盒提取细菌基因组,提取的核酸使用qubitdsdnahsassaykit进行定量并根据基因组大小计算基因组拷贝数。用天根微量样本dna提取试剂盒dp-316进行血液dna的提取并进行定量。将相当于10000拷贝的金黄色葡萄球菌核酸加入到10ng的人源核酸中。
3.样本打断:样品打断方法为
4.片段化的dna进行文库构建和测序分析
(1)dna片段末端修复
补平5’端缺口并连接磷酸基团,去除3’端的粘性末端。反应体系如下表3所示:
表3
(2)末端修复产物纯化
采用1×agencourtampurexp磁珠进行纯化,操作流程按照ampurexpbeads纯化说明书进行。
(3)连接接头标签序列
通过t4连接酶与平末端的dna片段相连;接头为含有标签序列的平末端接头,序列为:5’-ccatctcatccctgcgtgtctccgactcagnnnnnnnnncgat-3’(seqidno:5),5’-atcgnnnnnnnnnctgagtcggagacacgcagggatgagatgg/thiol-c6s-s/t/thiol-c6s-s/t-3’(seqidno:6),其中的nnnnnnnnn为标签序列。
反应体系如下表4所示:
表4
(4)连接产物纯化
采用1×agencourtampurexp磁珠进行纯化,操作流程按照说明书进行。纯化后将8个样本合并到一管中用真空离心浓缩仪将样本浓缩至41.5μl。
(5)探针捕获、延伸
探针捕获采用专门设计的捕获特异微生物序列的探针(表2所示的序列)与样本进行杂交并用phusiondna聚合酶进行延伸。探针5’端连接的测序平台公共接头序列为:5’-tccgctttcctctctatgggcagtcggtgat-3’(seqidno:7)。
反应体系如下表5所示:
表5
(6)捕获产物纯化
采用0.8xagencourtampurexp磁珠进行纯化,纯化两次。
(7)pcr扩增、纯化、检测浓度
pcr扩增采用platinumtmpfxdnapolymerase试剂盒,扩增10个循环,引物序列为:
t_pcr_a:5'-ccatctcatccctgcgtgtc-3’(seqidno:8);
plamp:5'-ccactacgcctccgctttcctctctatg-3'(seqidno:9)。
反应体系如下表6所示:
表6
检测浓度采用qubitdsdnahsassaykit2.0fluorometer试剂盒。
(8)检测文库质量
采用agilent2100bioanalyzer检测仪,操作流程按照agilent2100bioanalyzer说明书进行,agilent2100检测结果如图2所示。
(9)bgiseq-100测序
按照bgiseq-100使用说明书进行操作。
下机数据与金黄色葡萄球菌的待检测目标区域比对,将仅能比上金黄色葡萄球菌目标区域的读长(reads)进行计数,统计阴性样本与阳性样本中金黄色葡萄球菌不同探针捕获的reads情况(如图3),结果显示单引物探针捕获建库的测序结果可以明确判定阳性样本的检出。
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
sequencelisting
<110>深圳华大基因股份有限公司
<120>一种基于单引物探针捕获检测微生物目标序列的方法
<130>p2017-1-0139.cn
<160>9
<170>patentinversion3.3
<210>1
<211>36
<212>dna
<213>人工序列
<400>1
caaataaagatacgttatatagaacagtttggtctg36
<210>2
<211>36
<212>dna
<213>人工序列
<400>2
ctgttctaaatctattatttcgtcacattgatttgc36
<210>3
<211>36
<212>dna
<213>人工序列
<400>3
cgcgctgtttacttgattagctgcagctgttacttc36
<210>4
<211>36
<212>dna
<213>人工序列
<400>4
aacaaaaagatgcattaaaagcacaagctaatggtg36
<210>5
<211>43
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(31)..(39)
<223>nisa,c,g,ort
<400>5
ccatctcatccctgcgtgtctccgactcagnnnnnnnnncgat43
<210>6
<211>43
<212>dna
<213>人工序列
<220>
<221>misc_feature
<222>(5)..(13)
<223>nisa,c,g,ort
<400>6
atcgnnnnnnnnnctgagtcggagacacgcagggatgagatgg43
<210>7
<211>31
<212>dna
<213>人工序列
<400>7
tccgctttcctctctatgggcagtcggtgat31
<210>8
<211>20
<212>dna
<213>人工序列
<400>8
ccatctcatccctgcgtgtc20
<210>9
<211>28
<212>dna
<213>人工序列
<400>9
ccactacgcctccgctttcctctctatg28
- 还没有人留言评论。精彩留言会获得点赞!
- 用于检测CYP2C19基因多态性的引物、探针及检测试剂盒的制造方法与工艺
- 分子信标探针及引物对、GC基因SNPs位点检测方法与流程
- 一种表面修饰分子印迹固相微萃取探针及其制备与应用的制造方法与工艺
- 一种木瑚菌属食用菌鉴别用分子标记及引物和探针的制造方法与工艺
- 一种用于检测样品中牛凸隆病毒的荧光RT-PCR引物、探针和试剂盒及检测方法与制造工艺
- CYP2C19基因多态性检测引物、探针及试剂盒的制造方法与工艺
- 检测CYP2C9和VKORC1基因多态性的引物、探针和试剂盒的制造方法与工艺
- 一种用于检验临床样本中活菌体的荧光PCR引物探针组及试剂盒的制造方法与工艺
- 含反义碱基的探针法实时荧光PCR的制造方法与工艺
- PCR反应管及包含其的固相载具的制造方法与工艺
- 一种用于评估皮肤抗过敏情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤防御情况的引物和探针及试剂盒的制作方法
- 一种荧光定量PCR检测猪delta冠状病毒的引物、探针、试剂盒及方法
- 一种快速区分HP-PRRS疫苗GDr180株与野毒株的引物、探针及方法
- 一种用于检测样品中猪凸隆病毒的荧光rt-pcr引物和探针和试剂盒及检测方法
- 一种阿胶中驴、马、牛和猪源性巢式荧光pcr检测引物、探针、试剂盒、检测方法及应用
- 一种用于评估皮肤防晒情况的引物和探针及试剂盒的制作方法
- 一种用于评估皮肤抗皱情况的引物和探针及试剂盒的制作方法
- 一种用于检测car-t转导效率的引物、探针和方法
- 用于微嵌合检测和个体识别的引物、探针、试剂盒及方法
- 一种木瑚菌属食用菌鉴别用分子标记及引物和探针的制造方法与工艺
- 一组用于驴源性的实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一组用于鸡源性实时荧光PCR检测的特异性引物和探针的制造方法与工艺
- 一种快速检测异鳞蛇鲭和棘鳞蛇鲭源性成分的方法与制造工艺
- 一种检测食管癌相关标志物的探针组合及其试剂盒的制造方法与工艺
- 一种检测皮肤癌相关标志物的探针组合及其试剂盒的制造方法与工艺
- 一种检测骨髓瘤相关标志物的探针组合及其试剂盒的制造方法与工艺
- 一种用于检测样品中牛凸隆病毒的荧光RT-PCR引物、探针和试剂盒及检测方法与制造工艺
- CYP2C19基因多态性检测引物、探针及试剂盒的制造方法与工艺
- 检测CYP2C9和VKORC1基因多态性的引物、探针和试剂盒的制造方法与工艺