本发明涉及生物材料的检测,具体地说,涉及一种基于crispr-cas13a的适用于生物样本中特异性核酸片段的快速检测方法。
背景技术:
随着人类医学发展,人们逐渐认识到从基因水平上对疾病进行研究和诊断,是实现个体化精准医疗这一医学概念的关键。例如,对肿瘤患者血中的循环肿瘤dna进行快速检测,并对其中存在的突变基因进行确定,不仅可以实现肿瘤的早期诊断及其突变基因谱的绘制,而且还能够对后续的靶向药物治疗提供依据,避免药物浪费和耐药产生;对病毒或细菌感染患者的生物样品中存在的病原体进行分离提取后,利用基因诊断的方法对其基因进行快速鉴定,同样可以实现早期诊断和指导治疗。然而,这些在基因水平上进行的疾病诊断,都需要一种能够快速准确并且灵敏度高的核酸检测技术,在某些不发达地区或条件下,这种检测技术还需要简便易操作。
目前,医院和检测机构常用的核酸检测技术是基于pcr(聚合酶链式反应)技术发展而来的,其中最常用的就是将实时荧光定量pcr(realtime-pcr)和扩增阻碍突变pcr(arms-pcr)两种技术结合起来的实时荧光定量扩增阻碍突变pcr(realtime-arms-pcr)。realtime-pcr技术是由传统pcr技术并结合光谱技术发展而来的,通过在pcr反应体系中加入核酸嵌入荧光染料或者特异性淬灭探针,这两种荧光探针的信号都会随着核酸扩增而呈现线性增强,再通过pcr仪自带的荧光检测装置发出激发光并收集检测荧光信号,就可以实时反映pcr过程中每个循环的扩增水平;arms-pcr则是利用了taqdna聚合酶缺乏3’到5’的外切酶活性,在一定条件下pcr引物3’端的错配会导致扩增受到阻碍,产物急剧减少,因此针对不同的已知基因突变,设计适当的引物,就可以通过这种pcr方法区分出突变型和野生型基因。将这两种技术结合起来的realtime-arms-pcr,实现了特定基因片段的实时定量快速检测。
国内外已有多种基于realtime-arms-pcr技术的基因检测试剂盒应用于临床检测中,然而,现有技术仍具有诸多不足之处:1.pcr技术需要精确的温度系统控制,在一些不发达地区无法实现;2.检测灵敏度和特异性仍需进一步提高;3.pcr技术只能用于dna片段检测,rna片段需要提前加入一步逆转录的过程,操作复杂,而且开管操作容易造成样品污染。
因此,亟需提供一种温度控制简单,且灵敏度、特异性高的特异性片段检测方法。
技术实现要素:
为了解决现有技术中存在的问题,本发明的目的是提供温度控制简单,且灵敏度、特异性高的特异性片段检测方法,并进一步解决检测过程中样品易被污染的问题。
为了实现本发明的目的,本发明提供一种基于crispr-cas13a的特异性核酸片段的检测方法。
第一方面,本发明所提供的方法包括如下步骤(反应):
(1)rpa技术对待检片段进行核酸扩增反应。
参与该反应的试剂为:rpa引物(0.48μm)、模板(待检片段)、rpa酶预混液(来自twistdx公司)、醋酸镁(14mm);
其中,rpa引物的长度一般不超过50个核苷酸,优选为20-35个核苷酸。rpa引物的上游引物,需要在5’端加入启动子序列,以用于扩增产物后续进行体外转录。
所述启动子可以选择t7、t3、sp6等,优选为t7启动子。
rpa的扩增产物中gc含量一般不小于30%,不大于70%,优选为40%-60%;长度一般不超过500个核苷酸,优选为100-200个核苷酸。
需要说明的是,本发明rpa反应的模板(待检片段)既可以是dna,也可以是rna;当模板是rna时,需使用用于rna的rpa酶预混液,以实现反转录功能。
(2)将上述扩增产物进行体外转录。
参与该反应的试剂为:上述rpa扩增产物(1μg)、ntp混合液(6.7mm)、基于启动子的rna聚合酶预混液(来自neb公司)。
(3)基于cas13a蛋白独特的靶向rna激活的rna酶活性,进行核酸序列检测。
参与该反应的试剂包括:上述体外转录产物rna,cas13a蛋白,向导rna(crrna,与cas13a蛋白摩尔浓度相同),缓冲液(根据不同细菌来源的cas13a蛋白进行调整),信号报告试剂。
其中,所述cas13a蛋白可以来自不同细菌,包括leptotrichiashahii(lshcas13a,用量为1μm),leptotrichiabuccalis(lbucas13a,用量为40nm),leptotrichiawadei(lwcas13a,用量为40nm)等。优选为lbucas13a。
所述缓冲液的浓度需要根据cas13a蛋白的来源进行调整,通常有以下两种选择:用于lshcas13a的缓冲液a(50mmtris-hclph7.5,200mmnacl,10mmmgcl2)和用于lbucas13a及lwcas13a的缓冲液a(20mmhepesph7.0,50mmkcl,5mmmgcl2)。
所述向导rna(crrna)的结构为5’-锚定序列-向导序列-3’。
锚定序列根据cas13a蛋白的来源不同,分为:针对lshcas13a的5’-ggccaccccaauaucgaaggggacuaaaac-3’,针对lbucas13a的5’-gaccaccccaaaaaugaaggggacuaaaac-3’和针对lwcas13a的5’-gauuuagacuaccccaaaaacgaaggggacuaaaac-3’;
向导序列则与靶向rna中的片段匹配,长度为21-28个核苷酸,优选为28个。
进一步地,所述cas13a蛋白可以耐受一个碱基的错配,因此本发明用于检测单碱基突变核酸片段时,通常需要人为地在crrna的向导序列中提前加入一个错配,错配位置一般选择为突变碱基的3’末端的第一到第五个碱基,优选为第二个碱基。
所述信号报告试剂的阳性信号有多种选择,包括荧光、吸光值、显色反应等,但信号报告的条件都是基于cas13a蛋白的rna酶活性。作为优选,所述阳性信号为荧光信号(一般选用来自thermoscientific公司的rnasealertv2),在反应体系中加入一条两端分别连接荧光物质和淬灭剂的rna,当cas13a蛋白在crrna的帮助下,识别具有靶向序列的靶向rna后,被激活的rna酶活性可以降解该带有信号的rna,从而释放荧光信号,实现检测。
以荧光信号为例,应按以下公式计算:
最终荧光信号f=∑【f实验组每个时间点-f实验组最初-(f阴性对照组每个时间点-f阴性对照组最初)】
其中,阴性对照组为每个实验组相对应的没有加crrna的阴性信号组。
本发明所提供的方法用于定性,因此,若最终荧光信号高于阴性样品100倍以上即可判断为阳性,1000倍以上判断为强阳性;若高于阴性样品10倍到100倍之间,可增大样品投入量再次检测。
进一步地,为了降低信号检测过程中的假阳性,在步骤(3)的反应体系中,还可引入rna酶抑制剂(来自neb公司),以及背景rna(提取自hek293ft细胞),以排除反应体系中cas13a蛋白以外的(不小心引入的)rna酶对检测结果的影响。
本发明上述方案,利用cas13a在识别靶向rna后激活的不受序列限制的rna酶活性,降解反应体系中加入的信号报告rna分子,实现最终的信号放大和读取。检测灵敏度可以达到10-18摩尔/升,即阿摩尔级别,特异性可以达到用于检测具有单碱基差异的核酸片段。
上述三个步骤(反应)分开进行,所需时间分别为20-40分钟、120分钟和60-120分钟,共计用时约5个小时左右。
且作为优选,当待检片段为dna时,第一步反应温度为40℃,其余两步为37℃。
因此,第二方面,本发明为了简化操作步骤,将上述三个步骤进行三合一,可将反应时间显著缩短为120分钟,在反应过程中每5分钟记录反应信号。
当待检片段为dna时,反应温度全程控制为37℃即可;当待检片段为dna时,反应温度全程控制为37~40℃。
本发明无需复杂的温度控制仪器或系统,可以通过将三步反应全部加入在一个反应体系中,进一步简化操作流程,并实现上述有益效果。
“三合一”后,本发明进一步提供一种优选的反应体系(50μl):
rpa引物(0.48μm)、rpa酶预混液29.5μl(来自twistdx公司)、醋酸镁(14mm)、模板(待检片段)、ntp混合液(2mm)、基于启动子的rna聚合酶预混液(2μl,来自neb公司),cas13a蛋白(浓度根据不同细菌来源的cas13a蛋白进行调整),向导rna(crrna,与cas13a蛋白摩尔浓度相同),rna酶抑制剂(3μl,来自neb公司),背景rna(200ng,提取自hek293ft细胞),缓冲液(根据不同细菌来源的cas13a蛋白进行调整),信号报告试剂2μl(一般选用来自thermoscientific公司的rnasealertv2,125nm)。
最终信号计算公式和三步反应分开进行时相同。
本发明涉及到的原料或试剂均为普通市售产品,涉及到的操作如无特殊说明均为本领域常规操作。
在符合本领域常识的基础上,上述各优选条件,可以相互组合,得到具体实施方式。
本发明的有益效果在于:
本发明公开了一种基于cripsr-cas13a的核酸检测技术,其最主要的机制是cas13a蛋白可以在向导rna的帮助下识别具有靶向序列的rna片段,随后被激活的不受序列限制的rna酶活性,通过在反应体系中加入rna链降解导致的信号报告分子,最终实现具有靶向序列的rna片段的信号识别。
本发明通过加入基于rpa反应的核酸扩增步骤,不仅使该检测技术的应用范围扩大到rna和dna都可以检测,同时明显提高了检测的灵敏度,可以检测出阿摩尔级别的核酸。
由于cas13a蛋白在识别靶向序列时,通常可以耐受一个碱基的错配,本发明通过提前在向导rna的序列中加入人为错配碱基,使本检测技术的特异性达到可以识别具有单碱基差异的核酸序列。
本发明公开的核酸检测技术与以往的基于pcr技术的检测技术不同,反应全程无需复杂的温度控制仪器或系统;通过将三步反应全部加入在一个反应体系中,进一步简化操作流程,可以在两个小时内检测出具有特异序列的核酸片段。
发明人选用kras野生型基因片段和kras-g12d突变型基因片段作为检测模型,按照发明公布的核酸检测技术检测了两段基因,可以检测出约50am的kras野生型基因片段,同时通过向导rna的设计,可以区分出一个碱基差异的dna核酸序列,即kras野生型基因片段和kras-g12d突变型基因片段。
附图说明
图1为基于crispr-cas13a的核酸检测技术的灵敏度测试。所用的模式目的核酸序列为kras基因中的一个片段,序列为5’-ctgctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatcc-3’。合成的crrna序列为5’-gaccaccccaaaaatgaaggggactaaaacccaccagctccaactaccacaagtttat-3’。实验结果显示,与对照组(样品为双蒸水)相比,本发明公开的基于crispr-cas13a的核酸检测技术可以检测出约50am的kras基因片段。
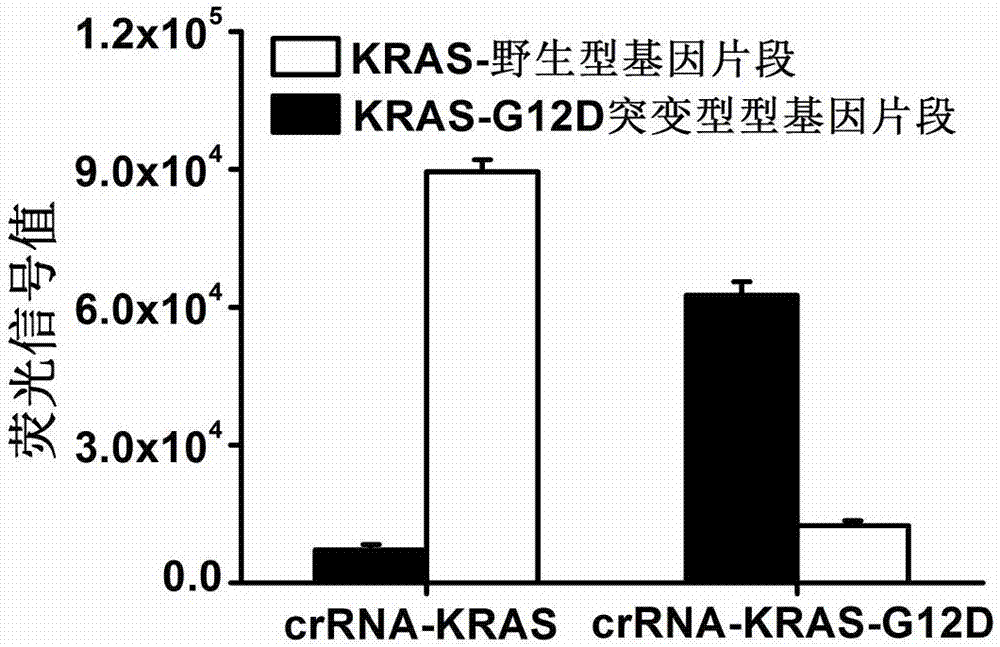
图2为基于crispr-cas13a的核酸检测技术的特异性测试。所用的模式目的核酸序列为kras野生型基因片段以及kras-g12d突变型基因片段,序列分别为5’-ctgctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatcc-3’和5’-ctgctgaaaatgactgaatataaacttgtggtagttggagctgatggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatcc-3’,其中大写a为突变碱基。合成的针对kras野生型基因片段的crrna-kras,序列为5’-gaccaccccaaaaatgaaggggactaaaacccacctgctccaactaccacaagtttat-3’;合成了针对kras突变型基因片段的crrna-kras-g12d,序列为5’-gaccaccccaaaaatgaaggggactaaaacccatctgctccaactaccacaagtttat-3’。实验结果显示,通过提前人为在crrna中加入错配的方式,本发明公开的基于crispr-cas13a的核酸检测技术可以区分出一个碱基差异的dna核酸序列,即kras野生型基因片段和kras-g12d突变型基因片段。
具体实施方式
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1
本实施例以kras基因,用于说明本发明检测方法(三合一)对目的基因的检测,并测试该方法的灵敏度。
首先,通过引物合成后退火的方法,合成kras基因中的一个片段作为检测的目的双链dna,序列为5’-ctgctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatcc-3’。
其次,合成rpa引物,上游引物为5’-taatacgactcactataggctgctgaaaatgactgaatataaactt-3’(其中5’端19个核苷酸为t7启动子),下游引物为5’-ggatcatattcgtccacaaaatgattct-3’。
再次,通过体外转录的方法,合成了针对kras基因片段的crrna,序列为5’-gaccaccccaaaaatgaaggggactaaaacccaccagctccaactaccacaagtttat-3’,其中大写字母为锚定序列,小写字母为向导序列,识别kras基因片段中的5’-ataaacttgtggtagttggagctggtgg-3’。
最后,构建50μl反应体系:
rpa引物(上下游引物各0.48μm);
rpa酶预混液(29.5μl,来自twistdx公司);
醋酸镁(14mm);
kras基因片段(按不同浓度稀释);
ntp混合液(2mm);
基于t7启动子的rna聚合酶预混液(2μl,来自neb公司);
lbucas13a蛋白(40nm);
针对kras基因片段的crrna(40nm);
rna酶抑制剂(3μl,来自neb公司);
背景rna(200ng,提取自hek293ft细胞);
缓冲液(20mmhepesph7.0,50mmkcl,5mmmgcl2);
rnasealertv22μl(125nm,来自thermoscientific公司)。
每个浓度的kras基因片段实验组应设置相应的不加crrna的阴性对照组。
将反应体系混匀后,放置入多功能酶标仪,设置反应条件为37℃,激发光波长488nm,反应时间为120分钟,在反应过程中每5分钟记录520nm处的荧光信号值,最终通过以下公式计算最终信号:
最终荧光信号f=∑【f实验组每个时间点-f实验组最初-(f阴性对照组每个时间点-f阴性对照组最初)】
阴性对照组为每个实验组相对应的没有加crrna的阴性信号组。
实验结果显示,与对照组(样品为双蒸水)相比,本发明公开的基于crispr-cas13a的核酸检测技术可以检测出约50am的kras基因片段(图1)。
实施例2
本实施例以kras基因及kras-g12d突变基因作为目的基因,用于说明本发明检测方法(三合一)对目的基因的检测,并测试该方法的特异性。
首先,通过引物合成后退火的方法,分别合成了kras基因片段以及kras-g12d突变片段作为检测的目的双链dna,序列分别为5’-ctgctgaaaatgactgaatataaacttgtggtagttggagctggtggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatcc-3’和5’-ctgctgaaaatgactgaatataaacttgtggtagttggagctgatggcgtaggcaagagtgccttgacgatacagctaattcagaatcattttgtggacgaatatgatcc-3’,其中大写a为突变碱基。
其次,合成了rpa引物,上游引物为5’-taatacgactcactataggctgctgaaaatgactgaatataaactt-3’(其中5’端19个核苷酸为t7启动子),下游引物为5’-ggatcatattcgtccacaaaatgattct-3’。
再次,由于cas13a蛋白普遍可以耐受一个碱基的错配,因此我们通过体外转录和提前加入人为错配的方法,合成了针对kras野生型基因片段的crrna-kras,序列为5’-gaccaccccaaaaatgaaggggactaaaacccacctgctccaactaccacaagtttat-3’,其中大写字母为锚定序列和人为加入的错配,小写字母为向导序列,识别kras野生型基因片段中的5’-ataaacttgtggtagttggagctggtgg-3’;合成了针对kras突变型基因片段的crrna-kras-g12d,序列为5’-gaccaccccaaaaatgaaggggactaaaacccatctgctccaactaccacaagtttat-3’,其中大写字母为锚定序列和人为加入的错配,小写字母为向导序列,识别kras-g12d突变型基因片段中的5’-ataaacttgtggtagttggagctgatgg-3’。
最后,构建50μl反应体系:
rpa引物(上下游引物各0.48μm);
rpa酶预混液(29.5μl,来自twistdx公司);
醋酸镁(14mm);
kras基因片段或kras-g12d突变片段(50nm);
ntp混合液(2mm);
基于t7启动子的rna聚合酶预混液(2μl,来自neb公司);
lbucas13a蛋白(40nm);
crrna-kras或crrna-kras-g12d(40nm);
rna酶抑制剂(3μl,来自neb公司);
背景rna(200ng,提取自hek293ft细胞);
缓冲液(20mmhepesph7.0,50mmkcl,5mmmgcl2);
rnasealertv22μl(125nm,来自thermoscientific公司);
设置相应的不加crrna的阴性对照组。
将反应体系混匀后,放置入多功能酶标仪,设置反应条件为37℃,激发光波长488nm,反应时间为120分钟,在反应过程中每5分钟记录520nm处的荧光信号值,最终通过以下公式计算最终信号:
最终荧光信号f=∑【f实验组每个时间点-f实验组最初-(f阴性对照组每个时间点-f阴性对照组最初)】
其中,阴性对照组为每个实验组相对应的没有加crrna的阴性信号组。
实验结果显示,通过提前人为在crrna中加入错配的方式,本发明公开的基于crispr-cas13a的核酸检测技术可以区分出一个碱基差异的dna核酸序列,即kras野生型基因片段和kras-g12d突变型基因片段(图2)。
应当理解的是,对上述实施例所用试剂或原料的用量进行等比例扩大或者缩小后的技术方案,与上述实施例的实质相同。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
序列表
<110>国家纳米科学中心
<120>一种基于crispr-cas13a的特异性核酸片段的检测方法
<160>8
<170>siposequencelisting1.0
<210>2
<211>30
<212>rna
<213>人工序列(artificialsequence)
<400>2
ggccaccccaauaucgaaggggacuaaaac30
<210>2
<211>30
<212>rna
<213>人工序列(artificialsequence)
<400>2
gaccaccccaaaaaugaaggggacuaaaac30
<210>3
<211>36
<212>rna
<213>人工序列(artificialsequence)
<400>3
gauuuagacuaccccaaaaacgaaggggacuaaaac36
<210>4
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>4
gaccaccccaaaaatgaaggggactaaaacccaccagctccaactaccacaagtttat58
<210>5
<211>58
<212>dna
<213>人工序列(artificialsequence)
<400>5
gaccaccccaaaaatgaaggggactaaaacccatctgctccaactaccacaagtttat58
<210>6
<211>46
<212>dna
<213>人工序列(artificialsequence)
<400>6
taatacgactcactataggctgctgaaaatgactgaatataaactt46
<210>7
<211>28
<212>dna
<213>人工序列(artificialsequence)
<400>7
ggatcatattcgtccacaaaatgattct28
<210>8
<211>28
<212>dna
<213>人工序列(artificialsequence)
<400>8
ataaacttgtggtagttggagctggtgg28