一种两步法合成2,6,8‑三甲基‑4‑壬醇的工艺方法与流程
本发明属于羟醛缩合以及催化加氢领域,具体涉及一种由甲基异丁基酮两步合成2,6,8-三甲基-4-壬醇的工艺方法。
背景技术:
脂肪醇是生产表面活性剂的主要原料,目前世界上大约90%的脂肪醇用于生产表面活性剂,约3%为直接应用,其它用途约占7%。洗涤剂醇(脂肪醇c12~c15)的衍生物——醇系表面活性剂因其去污力强、耐硬水、低温洗涤效果好、配伍能力强和生物降解快等综合性能优异得到了大力发展,从而导致了洗涤剂醇的需求量增加。其中,支链醇由于其特殊的性质,在日用化工上的应用特别受重视。支链醇与直链醇相比较,在碳数相同下,无色无味,而直链醇则有气味。支链醇对人体几乎无刺激性,而普通直链醇则有刺激性,特别是对皮肤过敏者。支链醇具有低凝固点,与相应的直链醇相比较,流动性、润湿性、渗透性都很优越。
我国高级醇生产工艺有两种路线:天然油脂路线和合成路线,前者以天然油脂为原料生产高级醇;后者以石油衍生物为原料制醇,资源丰富,原料价廉,其合成原料有乙烯、丙烯、正构烷烃和石蜡,生产方法有齐格勒法和羰基合成法、液蜡氧化法等。然而,对于长链高级醇,现有的方法存在反应条件苛刻、原料来源不易、生产成本过高等缺点。
2,6,8-三甲基-4-壬醇作为一种高度支链化的仲醇,具有低凝固点、流动性好、润湿性高及渗透性优越等优点,主要用于与环氧乙烷、聚乙二醇、酸等反应制备非离子表面活性剂。然而,目前还没有通过催化甲基异丁基酮两步合成2,6,8-三甲基-4-壬醇的工艺方法的相关报道。
技术实现要素:
为了提供一种成本低,转化率高,选择性好,绿色环保,且能够工业化生产的2,6,8-三甲基-4-壬醇的合成方法,本发明提供一种催化甲基异丁基酮两步合成2,6,8-三甲基-4-壬醇的工艺方法。第一步是催化甲基异丁基酮自缩合反应,第二步是自缩合产物的加氢还原。该方法具有催化剂制备简单,反应条件温和,产物收率高、副产物少,绿色环保等特点,具有较好的工业化前景。
本发明催化甲基异丁基酮“两步法”合成2,6,8-三甲基-4-壬醇的工艺如下式所示:
具体工艺方法步骤如下:
第一步、羟醛缩合反应:在间歇反应器中,以甲基异丁基酮为原料,在固体碱催化剂作用下,催化甲基异丁基酮自缩合反应生成自缩合产物;
第二步、加氢反应:在间歇反应器中,将第一步生成的自缩合产物在负载型加氢催化剂作用下,催化加氢还原合成2,6,8-三甲基-4-壬醇。
其中,第一步使用的固体碱催化剂为:ca(oh)2、mg(oh)2、碳酸氢钠、al2o3中的一种;
自缩合反应条件为:反应压力为:常压,反应温度为:85~110℃,反应时间为:12~20h,催化剂用量为:5~25wt%。
第二步使用m/al2o3作为加氢催化剂,该催化剂采用al2o3作为催化剂的载体,m作为催化剂的活性组分。
加氢还原反应条件为:反应温度为:80~150℃,反应压力为:1~5mpa,反应时间为:4h,反应搅拌速度为:600~1000r/min,催化剂用量为:1~5wt%。
第二步中加氢催化剂m/al2o3采用吸附沉淀法制备,具体工艺步骤如下:
(1)al2o3在使用之前在750℃焙烧4h。
(2)将活性组分的可溶性盐用去离子水配置成浸渍液,将al2o3置于浸渍液中,再用碱溶液调节ph值为8~11,静置后再经过洗涤、干燥和还原制得催化剂。
其中,步骤(2)所述的活性组分m为ru、ir、rh、pt、pd中的一种,m含量为1~5wt%,所述活性组分的可溶性盐为硝酸盐或碳酸盐或氯化物中的一种。
步骤(2)所述的碱溶液为naoh、koh、nahco3、k2co3、na2co3中的一种。
有益效果:
本发明催化甲基异丁基酮两步合成2,6,8-三甲基-4-壬醇的突出特点是:
(1)第一步的羟醛缩合反应操作简单,反应条件温和,产物收率高、副产物少。采用本发明的催化剂以及工艺条件,第一步甲基异丁基酮的转化率最高可达76%,选择性最高可达95%;
(2)采用本发明制备的m/al2o3催化剂,不仅有较高的催化活性,加氢还原转化率最高可达99%,同时选择性也高达99%。而且反应时间短,0.16mol的反应物4h即可完全转化为目标产物,催化剂用量少,催化剂用量与原料的质量比仅为2%。
附图说明
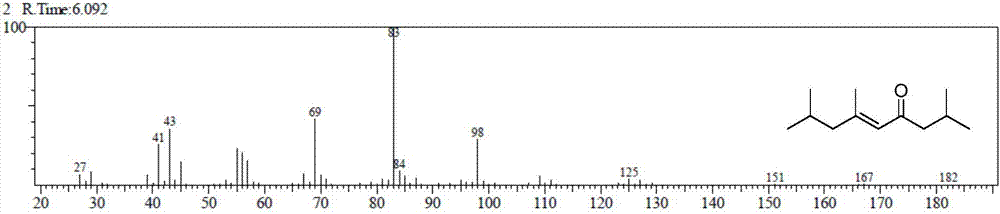
图1为实施例3催化甲基异丁基酮自缩合产物的气质谱图;
图2为实施例3催化甲基异丁基酮自缩合产物2的气质谱图;
图3为实施例3催化甲基异丁基酮自缩合产物4的气质谱图;
图4为实施例3合成的2,6,8-三甲基-4-壬醇的气质谱图。
具体实施方式
下面通过实施例对本发明作进一步的说明。
负载型加氢催化剂的制备方法如下:
实施例1
用γ-al2o3做载体,载体预先在750℃下恒温干燥4h。称取0.2730g的rucl3·xh2o溶于100g蒸馏水中,待完全溶解后得到浸渍液,将处理好的载体喷洒到浸渍液中,搅拌均匀,浸渍4h,然后加入1mol/lnaoh溶液调节ph值为8.0,持续搅拌4h,静置过夜;抽滤,去离子水洗涤至无氯离子,在110℃下干燥至恒重后,150℃氢气还原2h。其中活性组分ru占催化剂总质量的1.0%。
实施例2
用γ-al2o3做载体,载体预先在750℃下恒温干燥4h。称取0.583g的h2ircl6·xh2o溶于100g蒸馏水中,待完全溶解后得到浸渍液,将处理好的载体喷洒到浸渍液中,搅拌均匀,浸渍6h,然后加入1mol/lkoh溶液调节ph值为9.0,持续搅拌4h,静置过夜;抽滤,去离子水洗涤至无氯离子,在110℃下干燥至恒重后,200℃氢气还原2h。其中活性组分ir占催化剂总质量的2.0%。
实施例3
用γ-al2o3做载体,载体预先在750℃下恒温干燥4h。称取0.316g的rhcl3·3h2o溶于100g蒸馏水中,待完全溶解后得到浸渍液,将处理好的载体喷洒到浸渍液中,搅拌均匀,浸渍8h,然后加入1mol/lnahco3溶液调节ph值为9.5,持续搅拌4h,静置过夜;抽滤,去离子水洗涤至无氯离子,在110℃下干燥至恒重后,250℃氢气还原2h。其中活性组分rh占催化剂总质量的3.0%。
实施例4
用γ-al2o3做载体,载体预先在750℃下恒温干燥4h。称取1.111g的h2ptcl6·6h2o溶于100g蒸馏水中,待完全溶解后得到浸渍液,将处理好的载体喷洒到浸渍液中,搅拌均匀,浸渍10h,然后加入1mol/lk2co3溶液调节ph值为10.0,持续搅拌4h,静置过夜;抽滤,去离子水洗涤至无氯离子,在110℃下干燥至恒重后,300℃氢气还原2h。其中活性组分pt占催化剂总质量的4.0%。
实施例5
用γ-al2o3做载体,载体预先在750℃下恒温干燥4h。称取0.877g的pdcl2溶于100g蒸馏水中,待完全溶解后得到浸渍液,将处理好的载体喷洒到浸渍液中,搅拌均匀,浸渍12h,然后加入1mol/lna2co3溶液调节ph值为11.0,持续搅拌4h,静置过夜;抽滤,去离子水洗涤至无氯离子,在110℃下干燥至恒重后,400℃氢气还原2h。其中活性组分pd占催化剂总质量的5.0%。
2,6,8-三甲基-4-壬醇的合成方法如下:
实施例1
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取10g(5wt%)的mg(oh)2作为催化剂,常压下进行反应,反应温度85℃,反应时间12h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为56.12%,选择性为82.47%,缩合产物的收率为46.28%。
第二步、加氢还原反应:取加氢催化剂制备方法中实施例1制得的催化剂0.3g(1.0wt%)与30g第一步生成的自缩合产物,在反应温度80℃,1mpa氢气压力,搅拌速度1000r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为99.12%,选择性为86.08%,产物的收率为85.32%。
实施例2
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取20g(10wt%)的mg(oh)2作为催化剂,常压下进行反应,反应温度90℃,反应时间14h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为50.12%,选择性为98.74%,缩合产物的收率为49.49%。
第二步、加氢还原反应:取加氢催化剂制备方法中实施例2制得的催化剂0.45g(1.5wt%)与30g第一步生成的自缩合产物,在反应温度100℃,2mpa氢气压力,搅拌速度900r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为99.82%,选择性为89.99%,产物的收率为89.83%。
实施例3
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取30g(15wt%)的ca(oh)2作为催化剂,常压下进行反应,反应温度95℃,反应时间16h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为76.12%,选择性为95.04%,缩合产物的收率为72.34%。自缩合产物的气质谱图如图1、图2、图3所示,其中,图1中的缩合产物1,2,3和4为同分异构体。以化合物2,4为例,由图2和3可以看出,化合物相对分子质量均为182,表明自缩合产物为不饱和的酮。
第二步、加氢还原反应:取加氢催化剂制备方法中实施例3制得的催化剂0.6g(2.0wt%)与30g第一步生成的自缩合产物,在反应温度120℃,3mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为99.82%,选择性为99.03%,产物的收率为98.85%。合成的2,6,8-三甲基-4-壬醇的气质谱图如图4中所示,其中,还原产物1和2为顺反异构体。由质谱图分析可知,其相对分子质量均为186,产物为2,6,8-三甲基-4-壬醇。
实施例4
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取40g(20wt%)的碳酸氢钠作为催化剂,常压下进行反应,反应温度100℃,反应时间18h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为46.61%,选择性为97.34%,缩合产物的收率为45.37%。
第二步、加氢还原反应:取加氢催化剂制备方法中实施例4制得的催化剂0.75g(2.5wt%)与30g第一步生成的自缩合产物,在反应温度140℃,4mpa氢气压力,搅拌速度700r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为99.17%,选择性为99.23%,产物的收率为98.41%。
实施例5
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取50g(25wt%)的al2o3作为催化剂,常压下进行反应,反应温度110℃,反应时间20h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为73.80%,选择性为92.91%,缩合产物的收率为68.57%。
第二步、加氢还原反应:取加氢催化剂制备方法中实施例5制得的催化剂0.9g(2.0wt%)与30g第一步生成的自缩合产物,在反应温度150℃,5mpa氢气压力,搅拌速度600r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为99.47%,选择性为99.36%,产物的收率为98.83%。
对比实施例1
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取5g(2.5wt%)的ca(oh)2作为催化剂,常压下进行反应,反应温度95℃,反应时间16h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为5.64%,选择性为89.46%,缩合产物的收率为5.05%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于催化剂用量较少,催化剂所能提供的活性位不够,所以甲基异丁基酮转化率较低。
第二步、加氢还原反应:取加氢催化剂制备方法的实施例3制得的催化剂0.15g(0.5wt%)与30g第一步生成的自缩合产物,在反应温度120℃,3mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为22.23%,选择性为10.26%,产物的收率为2.28%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于催化剂用量较少,催化剂所能提供的活性位不够,导致反应物中的双键和羰基不能够被充分还原,所以产物2,6,8-三甲基-4-壬醇的收率较低。
对比实施例2
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取30g(15wt%)的ba(oh)2作为催化剂,常压下进行反应,反应温度95℃,反应时间16h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为24.22%,选择性为89.45%,缩合产物的收率为21.66%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,选用ba(oh)2时,由于催化剂ba(oh)2的碱性较弱,所能提供的碱性位不足,不利于缩合反应的进行,所以甲基异丁基酮转化率较低。
第二步、加氢还原反应:以ni/al2o3作为催化剂0.6g与30g第一步生成的自缩合产物,在反应温度120℃,3mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为88.45%,选择性为17.86%,产物的收率为15.80%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,选用ni/al2o3作为催化剂,由于催化剂中的活性位为ni,而ni的催化活性相对于rh较弱,所以反应物中的双键更易被还原,而反应物中的羰基较难被还原,因此产物2,6,8-三甲基-4-壬醇的选择性较低。
对比实施例3
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取30g(15wt%)的ca(oh)2作为催化剂,常压下进行反应,反应温度50℃,反应时间16h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为1.28%,选择性为89.95%,缩合产物的收率为1.15%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于在低温下,甲基异丁基酮的反应活性不高,原料不能充分反应,甲基异丁基酮的转化率低,因此,需要升高温度才能有利于促进反应物的转化。
第二步、加氢还原反应:取加氢催化剂制备方法的实施例3制得的催化剂0.6g与30g第一步生成的自缩合产物,在反应温度50℃,3mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为52.02%,选择性为55.92%,产物的收率为29.09%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于在低温下,反应物的活性不高,原料不能充分反应,产物2,6,8-三甲基-4-壬醇的收率较低,因此,需要升高温度才能有利于促进反应物的转化。
对比实施例4
第一步、羟醛缩合反应:在间歇反应器中,以200g甲基异丁基酮为原料,取30g(15wt%)的ca(oh)2作为催化剂,常压下进行反应,反应温度95℃,反应时间2h,得到甲基异丁基酮自缩合产物;采用气相色谱检测产物组成,其转化率为1.12%,选择性为99.04%,缩合产物的收率为1.11%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于反应时间太短,原料不能够完全反应,所以甲基异丁基酮转化率较低,而延长反应时间则可以得到较高的转化率和收率。
第二步、加氢还原反应:取加氢催化剂制备方法的实施例3制得的催化剂0.6g与30g第一步生成的自缩合产物,在反应温度120℃,3mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应1h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为87.56%,选择性为10.89%,产物的收率为9.54%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于反应时间太短,原料不能够完全反应,所以2,6,8-三甲基-4-壬醇的收率较低,而延长反应时间则可以得到较高的收率。
对比实施例5
第二步、加氢还原反应:取加氢催化剂制备方法的实施例3制得的催化剂0.6g与30g第一步生成的自缩合产物,在反应温度120℃,0.1mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为9.28%,选择性为1.33%,产物的收率为0.12%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,由于反应压力较低,则反应物中氢气浓度较低,不利于反应物的转化,所以2,6,8-三甲基-4-壬醇的收率较低,而增加反应压力则可以得到较高的收率。
对比实施例6
第二步、加氢还原反应:以非负载型的骨架钌作为催化剂0.6g与30g第一步生成的自缩合产物,在反应温度120℃,3mpa氢气压力,搅拌速度800r/min的条件下,进行加氢反应4h,合成2,6,8-三甲基-4-壬醇。采用气相色谱检测产物组成,其转化率为20.65%,选择性为9.89%,产物的收率为2.04%。
与2,6,8-三甲基-4-壬醇合成方法中的实施例3相比较,载体对产物的收率影响较大,相较于选用氧化铝作为载体的ru/al2o3催化剂,选用无载体的骨架钌作为催化剂,产物2,6,8-三甲基-4-壬醇的选择性和收率较低,可能是因为反应过程中骨架钌易团聚,从而降低了催化剂的稳定性,使得反应的转化率降低。而选用ru/al2o3催化剂,由于ru在氧化铝中分散较为均匀,没有发生团聚现象,从而提高了催化剂的稳定性,有利于提高原料的转化率,使得产物的收率也明显提高,因此,载体的存在可以明显提高产物收率。
以上所述,仅为本发明较佳的具体实施方案,本发明的保护范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见得到的技术方案的简单变化或等效替换均落在本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!