硫醚化吲哚类化合物的制备方法与流程
本发明属于有机合成技术领域,具体来说涉及一种硫醚化吲哚类化合物的制备方法。
背景技术:
碳-硫键的形成反应在有机硫化合物的合成中起到了非常关键的作用,而有机硫化合物又是许多药物分子或一些材料分子的重要片段和重要结构单元。因此,近年来对碳-硫键构建新方法的探索已引起了许多科学家的关注。目前构建碳-硫键主要方法是利用过渡金属催化,该方法需要用昂贵和对空气敏感的金属催化剂,且硫源恶臭、不稳定、具有挥发性。
技术实现要素:
针对现有技术的不足,本发明的目的在于提供一种硫醚化吲哚类化合物的制备方法,该制备方以取代苯基磺酰氯为母体使用一锅法制备硫醚化吲哚类化合物。
本发明的目的是通过下述技术方案予以实现的。
一种硫醚化吲哚类化合物的制备方法,按照下述步骤进行:
将催化剂、吲哚类反应物、水合肼、取代苯基磺酰氯和溶剂在密闭容器中混合并于70~100℃下搅拌反应13~17小时,得到硫醚化吲哚类化合物,其中,所述吲哚类反应物、水合肼和取代苯基磺酰氯的物质的量的比为1:1:2,所述催化剂为碘单质,所述溶剂为1,4-二氧六环,所述吲哚类反应物的结构通式为:
所述取代苯基磺酰氯的结构通式为:
其中,所述r1为氢、氟、氯、溴、硝基、甲基或甲氧基;所述r2为氢或甲基,所述r3为氢、甲基或苯基,所述r4为甲基或氯。
在上述技术方案中,所述取代苯基磺酰氯为对甲苯磺酰氯、邻甲苯磺酰氯或间氯苯磺酰氯。
在上述技术方案中,用体积比为(3~50):1的石油醚和乙酸乙酯的混合物作为流动相对搅拌反应后得到的硫醚化吲哚类化合物进行梯度洗脱,得到固体的硫醚化吲哚类化合物。
在上述技术方案中,所述搅拌反应的时间通过薄层色谱法确定。
在上述技术方案中,所述催化剂与吲哚类反应物的物质的量的比为1:10。
在上述技术方案中,所述吲哚类反应物在所述溶剂中的浓度为1~1.5mol/l。
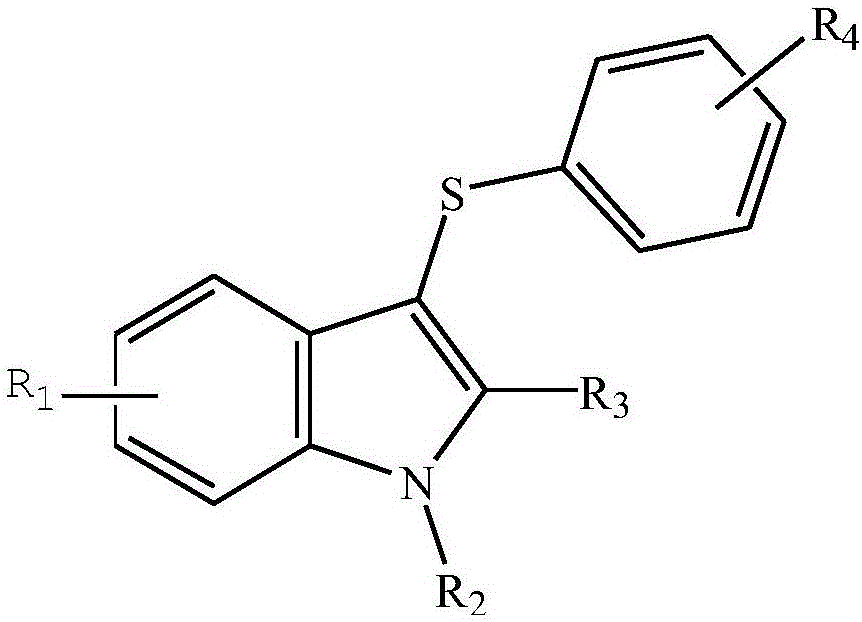
一种上述制备方法得到的硫醚化吲哚类化合物,所述硫醚化吲哚类化合物的结构通式为:
其中,所述r1为氢、氟、氯、溴、硝基、甲基或甲氧基;所述r2为氢或甲基,所述r3为氢、甲基或苯基,所述r4为甲基或氯。
一种上述制备方法在合成硫醚化吲哚类化合物中的应用,产率为41~86%。
相比于现有技术,本发明的制备方法的有益效果为:
1.原料和催化剂便宜易得。
2.本实验采用一锅法制得产物,步骤简单,副产物少。
附图说明
图1为本发明的硫醚化吲哚类化合物的结构通式。
具体实施方式
本发明的制备方法的反应机理为:
首先取代苯基磺酰氯在水合肼存在下,原位地生成中间体磺酰肼
吲哚类反应物为吲哚或吲哚类化合物,应用吲哚类反应物、取代苯基磺酰氯与水合肼在碘的催化下进行串联反应,合成出一系列的硫醚化吲哚类化合物。
本发明的制备方法能够合成的典型的硫醚化吲哚类化合物为:
1、3-[(4-甲苯基)巯基]-吲哚;
2、1,2-二甲基-3-[(4-甲苯基)巯基]-吲哚;
3、2-甲基-3-[(4-甲苯基)巯基]-吲哚;
4、2-苯基-3-[(4-甲苯基)巯基]-吲哚;
5、4-甲氧基-3-[(4-甲苯基)巯基]-吲哚;
6、5-甲基-3-[(4-甲苯基)巯基]-吲哚;
7、5-氟-3-[(4-甲苯基)巯基]-吲哚;
8、5-氯-3-[(4-甲苯基)巯基]-吲哚;
9、5-溴-3-[(4-甲苯基)巯基]-吲哚;
10、5-硝基-3-[(4-甲苯基)巯基]-吲哚;
11、6-氟-3-[(4-甲苯基)巯基]-吲哚;
12、6-氯-3-[(4-甲苯基)巯基]-吲哚;
13、6-甲基-3-[(4-甲苯基)巯基]-吲哚;
14、3-[(2-甲苯基)巯基]-吲哚;
15、3-[(3-氯苯基)巯基]-吲哚;
下面结合具体实施例进一步说明本发明的技术方案。
下述实施例合成的硫醚化吲哚类化合物的命名与结构如表1所示:
表1硫醚化吲哚类化合物的命名与结构
表1合成用到的所有吲哚类反应物,其纯度均为98%,购买厂家:萨恩化学技术(上海)有限公司。表1合成用到的所有取代苯基磺酰氯,其纯度也都为98%,购买厂家:萨恩化学技术(上海)有限公司。
核磁共振的仪器以及型号为核磁共振光谱(1hnmr)测试仪,bruckerarx400(400mhz),化学位移使用四甲基硅烷作为内标。
下述产率测定方法:首先称量出产物的质量,用产物的质量除以产物的摩尔质量,得到产物的物质的量。然后用产物的物质的量除以原料中吲哚类反应物的物质的量,就得到该产物的产率。
实施例1
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测,待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(81.7mg,产率:68%).经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.36(s,1h),7.61(dd,j=7.9,1.1hz,1h),7.48(d,j=2.6hz,1h),7.43(d,j=8.0hz,1h),7.29–7.24(m,1h),7.15(td,j=7.5,7.0,1.0hz,1h),7.06–7.00(m,2h),6.97(d,j=8.1hz,2h),2.24(s,3h)
实施例2:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将1,2-二甲基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=50:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(68.1mg,产率:51%).经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ7.57(d,j=7.8hz,1h),7.31(s,1h),7.20(t,j=7.6hz,1h),7.11(t,j=7.4hz,1h),6.93(s,4h),3.71(s,3h),2.49(s,3h),2.22(s,3h).
实施例3:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将2-甲基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(68.1mg,产率:51%)。经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.21(s,1h),7.54(d,j=7.7hz,1h),7.34(d,j=8.0hz,1h),7.21–7.15(m,1h),7.15–7.09(m,1h),6.96(s,4h),2.52(s,3h),2.24(s,3h).
实施例4:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将2-苯基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于70℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=50:1洗脱),得到褐色吲哚芳基硫醚固体产物(121.2mg,产率:77%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.40(s,1h),7.71(d,j=7.4hz,1h),7.62(d,j=7.8hz,1h),7.44–7.30(m,5h),7.24–7.17(m,1h),7.14(t,j=7.4hz,1h),7.01–6.93(m,4h),2.21(s,3h).
实施例5:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将4-甲氧基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。17小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(54.9mg,产率:41%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.30(s,1h),7.18(d,j=2.0hz,1h),7.13(d,j=8.0hz,1h),7.10(d,j=1.8hz,1h),7.09(s,1h),6.99(s,1h),6.97(s,1h),6.95(s,1h),6.52(d,j=7.8hz,1h),3.71(s,3h),2.24(s,3h).
实施例6:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将5-甲基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡粉色吲哚芳基硫醚固体产物(91.0mg,产率:72%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.28(s,1h),7.43(d,j=2.5hz,1h),7.41(s,1h),7.32(d,j=8.3hz,1h),7.08(d,j=8.0hz,1h),7.00(q,j=8.2hz,4h),2.41(s,3h),2.25(s,3h).
实施例7:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将5-氟吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=3:1洗脱),得到黄色吲哚芳基硫醚固体产物(83.3mg,产率:65%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.33(s,1h),7.48(s,1h),7.32(dd,j=8.7,3.8hz,1h),7.24(s,1h),7.03(s,1h),7.00(d,j=7.1hz,4h),2.25(s,3h).
实施例8:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将5-氯吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=5:1洗脱),得到土黄色吲哚芳基硫醚固体产物(92.0mg,产率:67%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.36(s,1h),7.58(s,1h),7.45(s,1h),7.31(d,j=8.6hz,1h),7.19(d,j=8.6hz,1h),7.01(m,4h),2.25(s,3h).
实施例9:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将5-溴吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=3:1洗脱),得到土黄色吲哚芳基硫醚固体产物(109.4mg,产率:69%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.37(s,1h),7.75(s,1h),7.44(s,1h),7.33(d,j=8.6hz,1h),7.27(d,j=8.6hz,1h),7.00(m,4h),2.25(s,3h).
实施例10:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将5-硝基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于100℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到橙黄色吲哚芳基硫醚固体产物(105.9mg,产率:75%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.99(s,1h),8.56(s,1h),8.15(d,j=8.9hz,1h),7.64(s,1h),7.48(d,j=9.0hz,1h),7.07–6.98(m,4h),2.26(s,3h).
实施例11:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将6-氟吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(80.6mg,产率:63%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.30(s,1h),7.49(dd,j=8.6,5.4hz,1h),7.41(d,j=2.1hz,1h),7.08(d,j=9.3hz,1h),7.00(q,j=8.2hz,4h),6.90(t,j=9.1hz,1h),2.25(s,3h).
实施例12:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将6-氯吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(117.3mg,产率:86%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.32(s,1h),7.49(d,j=8.5hz,1h),7.46–7.43(m,1h),7.40(s,1h),7.11(d,j=8.4hz,1h),7.02–6.97(m,4h),2.25(s,3h).
实施例13:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将7-甲基吲哚(0.5mmol)和对甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。14小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到黄色吲哚芳基硫醚固体产物(73.2mg,产率:58%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.30(s,1h),7.48(d,j=2.7hz,1h),7.47–7.43(m,1h),7.05(dd,j=17.4,7.6hz,4h),6.96(d,j=8.1hz,2h),2.53(s,3h),2.24(s,3h).
实施例14:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将吲哚(0.5mmol)和邻甲苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环,水合肼(1mmol),碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。13小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡粉色吲哚芳基硫醚固体产物(83.6mg,产率:70%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.42(s,1h),7.58(d,j=7.9hz,1h),7.48(d,j=2.6hz,1h),7.45(d,j=8.2hz,1h),7.28(d,j=7.3hz,1h),7.17(d,j=7.3hz,1h),7.15–7.11(m,1h),7.00–6.94(m,1h),6.89(t,j=7.5hz,1h),6.71(d,j=7.8hz,1h),2.49(s,3h).
实施例15:
合成硫醚化吲哚类化合物的结构式为:
该硫醚化吲哚类化合物的制备方法按照下述步骤予以进行:
将吲哚(0.5mmol)和间氯苯磺酰氯(0.5mmol)加入到15ml耐压瓶中,然后依次加入0.5ml1,4-二氧六环、水合肼(1mmol)和碘单质(0.05mmol)。旋紧耐压瓶封口,于90℃下充分搅拌反应。17小时后,用薄层色谱法检测。待反应完毕后,停止加热。此时,将体系转移至10ml圆底烧瓶,减压蒸发旋干溶剂,得到待过柱分离物。然后用硅胶-柱色谱将产物分离出来(用石油醚:乙酸乙酯=10:1洗脱),得到淡黄色吲哚芳基硫醚固体产物(94.3mg,产率:73%)经核磁1hnmr测定,1hnmr(400mhz,cdcl3)δ8.45(s,1h),7.59(d,j=7.9hz,1h),7.50(d,j=2.6hz,1h),7.46(d,j=8.2hz,1h),7.31–7.26(m,1h),7.21–7.16(m,1h),7.08(d,j=7.8hz,1h),7.05(d,j=1.8hz,1h),7.01(dt,j=7.7,1.5hz,1h),6.97(dt,j=7.6,1.5hz,1h).
上述实施例合成硫醚化吲哚类化合物所涉及的吲哚类反应物和取代苯基磺酰氯的名称以及结构式如下:
吲哚
对甲苯磺酰氯
1,2-二甲基吲哚
2-甲基吲哚
2-苯基吲哚
4-甲氧基吲哚
5-甲基吲哚
5-氟吲哚
5-氯吲哚
5-溴吲哚
5-硝基吲哚
6-氟吲哚
6-氯吲哚
7-甲基吲哚
邻甲苯磺酰氯
间氯苯磺酰氯
在本发明的技术方案中,通过调节吲哚类反应物的类型、取代苯基磺酰氯、反应温度和反应时间均能够达到上述实施例一致的性质。
本专利得到的资助:本发明得到大学生创新创业项目基金(国家级)的资助(资助号:201610065004)。
以上对本发明做了示例性的描述,应该说明的是,在不脱离本发明的核心的情况下,任何简单的变形、修改或者其他本领域技术人员能够不花费创造性劳动的等同替换均落入本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!